| REACTIVITY | H |
Product Information
1. Label the appropriate number of PCR tubes or PCR plates compatible with the model of real-time PCR machine to be used. PCR reactions should be performed in duplicate and should include a tube with no DNA to control for contamination, and a serial dilution of a 2% total input chromatin DNA (undiluted, 1:5, 1:25, 1:125), which is used to create a standard curve and determine amplification efficiency.2. Add 2 μl of the appropriate ChIP DNA sample to each tube or well of the PCR plate.
3. Prepare a master PCR reaction mix as described below. Add enough reagents for two extra reactions to account for loss of volume. Add 18 μl of the master PCR reaction mix to each PCR reaction tube or well of the PCR plate.
Reagent Volume for 1 PCR Reaction (20 μl)
Nuclease-free H2O 6 μl
5 μM SimpleChIP® Primers 2 μl
2X SYBR® Green Reaction Mix 10 μl
4. Start the following PCR reaction program:
a. Initial Denaturation: 95°C for 3 min.
b. Denaturation: 95°C for 15 sec.c. Anneal and Extension: Primer-specific temp. for 60 sec.
d. Repeat steps b and c for a total of 40 cycles.
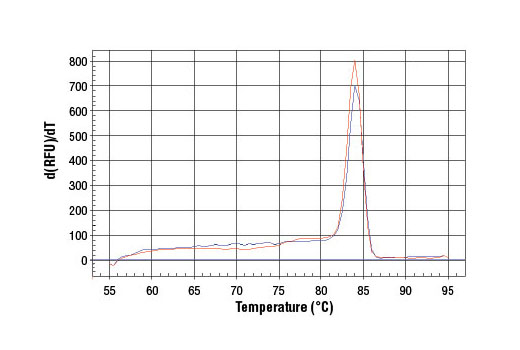
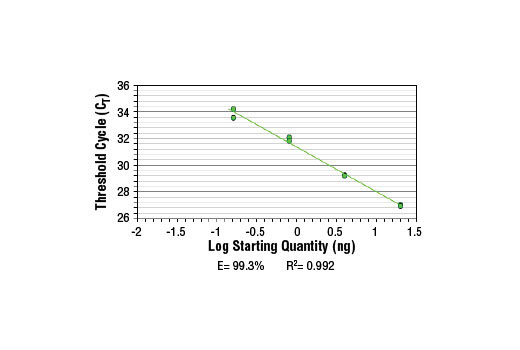
5. Analyze quantitative PCR results using software provided with the real-time PCR machine.
Specific for product: SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) #9005.
Reagents Included:
Reagents Not Included:
| ! | This ! signifies an important step in the protocol regarding volume changes based on the number of immunoprecipitation preparations (IP preps). One IP prep is defined as 4 x 106 tissue cultured cells or 25 mg or disaggregated tissue. |
| !! | This !! signifies an important step to dilute a buffer before proceeding. |
| SAFE STOP | This is a safe stopping point in the protocol, if stopping is necessary. |
When harvesting tissue, remove unwanted material such as fat and necrotic material from the sample. Tissue can then be processed and cross-linked immediately, or frozen on dry ice and stored at -80°C for processing later. For optimal chromatin yield and ChIP results, use 25 mg of tissue for each immunoprecipitation to be performed. The chromatin yield does vary between tissue types and some tissues may require more than 25 mg for each immunoprecipitation. Please see Appendix A for more information regarding the expected chromatin yield for different types of tissue. One additional chromatin sample should be processed for Analysis of Chromatin Digestion and Concentration (Section IV). If desired, five additional chromatin samples should be processed for Optimization of Chromatin Digestion (Appendix B).
(!) All buffer volumes should be increased proportionally based on the number of IP preps in the experiment.
For optimal ChIP results, use approximately 4 X 106 cells for each immunoprecipitation to be performed (at least 12 X 106 cells are required in order to include positive and negative controls). For HeLa cells, one IP is equivalent to half of a 15 cm culture dish containing cells that are 90% confluent in 20 ml of growth medium. One additional sample should be processed for Analysis of Chromatin Digestion and Concentration (Section IV). Since every cell type is different, we recommend including one extra dish of cells in experiment to be used for determination of cell number using a hemocytometer or cell counter.
(!) All buffer volumes should be increased proportionally based on the number of 15 cm tissue culture dishes (or 20 ml suspension cells) used.
(!) All buffer volumes should be increased proportionally based on the number of IP preps in the experiment.
(!!) IMPORTANT: Once in solution, store 1M DTT at -20°C.
NOTE: For optimal ChIP results, it is highly critical that the chromatin is of appropriate size and concentration. Over-digestion of chromatin may diminish signal in the PCR quantification. Under-digestion of chromatin may lead to increased background signal and lower resolution. Adding too little chromatin to the IP may result in diminished signal in the PCR quantification. A protocol for optimization of chromatin digestion can be found in Appendix B.
For optimal ChIP results, use approximately 5 to 10 µg of digested, cross-linked chromatin (as determined in Section IV) per immunoprecipitation. This should be roughly equivalent to a single 100 µl IP prep from 25 mg of disaggregated tissue or 4 x 106 tissue culture cells. Typically, 100 µl of digested chromatin is diluted into 400 µl 1X ChIP Buffer prior to the addition of antibodies. However, if more than 100 µl of chromatin is required per IP, the cross-linked chromatin preparation does not need to be diluted as described below. Antibodies can be added directly to the undiluted chromatin preparation for immunoprecipitation of chromatin complexes.
(!) All buffer volumes should be increased proportionally based on the number of immunoprecipitations in the experiment.
NOTE: Most antibodies from Cell Signaling Technology work optimally between 1 and 2 ug per IP sample. In the case where there are multiple samples with varying concentrations, it is best to match the negative control Normal Rabbit IgG #2729 to the highest antibody concentration.
(!) All buffer volumes should be increased proportionally based on the number of immunoprecipitations in the experiment.
| Primer length: | 24 nucleotides |
| Optimum Tm: | 60°C |
| Optimum GC: | 50% |
| Amplicon size: | 150 to 200 bp (for standard PCR) |
| 80 to 160 bp (for real-time quantitative PCR) |
| Reagent | Volume for 1 PCR Reaction (18 µl) |
|---|---|
| Nuclease-free H2O | 12.5 µl |
| 10X PCR Buffer | 2.0 µl |
| 4 mM dNTP Mix | 1.0 µl |
| 5 µM RPL30 Primers | 2.0 µl |
| Taq DNA Polymerase | 0.5 µl |
| a. | Initial Denaturation | 95°C | 5 min |
| b. | Denature | 95°C | 30 sec |
| c. | Anneal | 62°C | 30 sec |
| d. | Extension | 72°C | 30 sec |
| e. | Repeat Steps b-d for a total of 34 cycles. | ||
| f. | Final Extension | 72°C | 5 min |
| Reagent | Volume for 1 PCR Reaction (18 µl) |
|---|---|
| Nuclease-free H2O | 6 µl |
| 5 µM RPL30 Primers | 2 µl |
| SimpleChIP® Universal qPCR Master Mix #88989 | 10 µl |
| a. | Initial Denaturation | 95°C 3 min |
| b. | Denature | 95°C 15 sec |
| c. | Anneal and Extension: | 60°C 60 sec |
| d. | Repeat steps b and c for a total of 40 cycles. | |
Analyze quantitative PCR results using the software provided with the real-time PCR machine. Alternatively, one can calculate the IP efficiency manually using the Percent Input Method and the equation shown below. With this method, signals obtained from each immunoprecipitation are expressed as a percent of the total input chromatin.
Percent Input = 2% x 2(C[T] 2%Input Sample - C[T] IP Sample)
C[T] = CT = Threshold cycle of PCR reaction
The immuno-enriched DNA samples prepared with this kit are directly compatible with ChIP-seq. For downstream NG-sequencing DNA library construction, use a DNA library preparation protocol or kit compatible with your downstream sequencing platform. For sequencing on Illumina® platforms, we recommend DNA Library Prep Kit for Illumina® (ChIP-seq, CUT&RUN) #56795 and its associated index primers Multiplex Oligos for Illumina® (Single Index Primers) (ChIP-seq, CUT&RUN) #29580 or Multiplex Oligos for Illumina® (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538.
Recommendations:
When harvesting cross-linked chromatin from tissue samples, the yield of chromatin can vary significantly between tissue types. The table to the right provides a range for the expected yield of chromatin from 25 mg of tissue compared to 4 x 106 HeLa cells, and the expected DNA concentration, as determined in Section IV of the protocol. For each tissue type, disaggregation using a Medimachine (BD Biosciences) or a Dounce homogenizer yielded similar amounts of chromatin. However, chromatin processed from tissues disaggregated using the Medimachine typically gave higher IP efficiencies than chromatin processed from tissues disaggregated using a Dounce homogenizer. A Dounce homogenizer is strongly recommended for disaggregation of brain tissue, as the Medimachine does not adequately disaggregate brain tissue into a single-cell suspension. For optimal ChIP results, we recommend using 5 to 10 µg of digested, cross-linked chromatin per immunoprecipitation; therefore, some tissues may require harvesting more than 25 mg per each immunoprecipitation.
| Tissue/Cell | Total Chromatin Yield | Expected DNA Concentration |
|---|---|---|
| Spleen | 20-30 µg per 25 mg tissue | 200-300 µg/ml |
| Liver | 10-15 µg per 25 mg tissue | 100-150 µg/ml |
| Kidney | 8-10 µg per 25 mg tissue | 80-100 µg/ml |
| Brain | 2-5 µg per 25 mg tissue | 20-50 µg/ml |
| Heart | 2-5 µg per 25 mg tissue | 20-50 µg/ml |
| HeLa | 10-15 µg per 4 x 106 cells | 100-150 µg/ml |
Optimal conditions for the digestion of cross-linked chromatin DNA to 150-900 base pairs in length is highly dependent on the ratio of Micrococcal Nuclease to the amount of tissue or number of cells used in the digest. Below is a protocol for determination of the optimal digestion conditions for a specific tissue or cell type.
| Problem | Possible Causes | Recommendation |
|---|---|---|
| 1. Concentration of the digested chromatin is too low. | Not enough cells added to the chromatin digestion or nuclei were not completely lysed after digestion. | If DNA concentration of the chromatin preparation is close to 50 µg/ml, add additional chromatin to each IP to give at least 5 µg/IP and continue with protocol. Count a separate plate of cells before cross-linking to determine an accurate cell number and/or visualize nuclei under microscope before and after sonication to confirm complete lysis of nuclei. |
| 2. Chromatin is under-digested and fragments are too large (greater than 900 bp). | Cells may have been over cross-linked. Cross-linking for longer than 10 min may inhibit digestion of chromatin. Too many cells or not enough Micrococcal Nuclease was added to the chromatin digestion. | Perform a time course at a fixed formaldehyde concentration. Shorten the time of cross-linking to 10 min or less. Count a separate plate of cells before cross-linking to determine accurate cell number and see Appendix B for optimization of chromatin digestion. |
| 3. Chromatin is over-digested and fragments are too small (exclusively 150 bp mono-nucleosome length). Complete digestion of chromatin to mono-nucleosome length DNA may diminish signal during PCR quantification, especially for amplicons greater than 150 bp in length. | Not enough cells or too much Micrococcal Nuclease added to the chromatin digestion. | Count a separate plate of cells before cross-linking to determine accurate cell number and see Appendix B for optimization of chromatin digestion. |
| 4. No product or very little product in the input PCR reactions. | Not enough DNA added to the PCR reaction or conditions are not optimal. PCR amplified region may span nucleosome-free region. Not enough chromatin added to the IP or chromatin is over-digested. | Add more DNA to the PCR reaction or increase the number of amplification cycles. Optimize the PCR conditions for experimental primer set using purified DNA from cross-linked and digested chromatin. Design a different primer set and decrease length of amplicon to less than 150 bp (see primer design recommendations in Section VIII). For optimal ChIP results add 5-10 µg chromatin per IP. See recommendations for problems 1 and 3 above. |
| 5. No product in the positive control Histone H3-IP RPL30 PCR reaction. | Not enough chromatin or antibody added to the IP reaction or IP incubation time is too short. Incomplete elution of chromatin from Protein G beads. | Be sure to add 5-10 µg of chromatin and 10 µl of antibody to each IP reaction and incubate with antibody over-night and an additional 2 h after adding Protein G beads. Elution of chromatin from Protein G beads is optimal at 65°C with frequent mixing to keep beads suspended in solution. |
| 6. Quantity of product in the negative control Rabbit IgG-IP and positive control Histone H3-IP PCR reactions is equivalent. | Too much or not enough chromatin added to the IP reaction. Alternatively, too much antibody added to the IP reaction. Too much DNA added to the PCR reaction or too many cycles of amplification. | Add no more than 15 µg of chromatin and 10 µl of histone H3 antibody to each IP reaction. Reduce the amount of normal rabbit IgG to 1 µl per IP. Add less DNA to the PCR reaction or decrease the number of PCR cycles. It is very important that the PCR products are analyzed within the linear amplification phase of PCR. Otherwise, the differences in quantities of starting DNA can not be accurately measured. |
| 7. No product in the Experimental Antibody-IP PCR reaction. | Not enough DNA added to the PCR reaction. Not enough antibody added to the IP reaction. Antibody does not work for IP. | Add more DNA to the PCR reaction or increase the number of amplification cycles. Typically a range of 1 to 5 µg of antibody are added to the IP reaction; however, the exact amount depends greatly on the individual antibody. Increase the amount of antibody added to the IP. Find an alternate antibody source. |
posted December 2011
revised April 2022
Protocol Id: 82
Human
The chromatin immunoprecipitation (ChIP) assay is a powerful and versatile technique used for probing protein-DNA interactions within the natural chromatin context of the cell (1,2). This assay can be used to either identify multiple proteins associated with a specific region of the genome or to identify the many regions of the genome bound by a particular protein (3-6). ChIP can be used to determine the specific order of recruitment of various proteins to a gene promoter or to "measure" the relative amount of a particular histone modification across an entire gene locus (3,4). In addition to histone proteins, the ChIP assay can be used to analyze binding of transcription factors and co-factors, DNA replication factors, and DNA repair proteins. When performing the ChIP assay, cells are first fixed with formaldehyde, a reversible protein-DNA cross-linking agent that "preserves" the protein-DNA interactions occurring in the cell (1,2). Cells are lysed and chromatin is harvested and fragmented using either sonication or enzymatic digestion. Fragmented chromatin is then immunoprecipitated with antibodies specific to a particular protein or histone modification. Any DNA sequences that are associated with the protein or histone modification of interest will co-precipitate as part of the cross-linked chromatin complex and the relative amount of that DNA sequence will be enriched by the immunoselection process. After immunoprecipitation, the protein-DNA cross-links are reversed and the DNA is purified. Standard PCR or quantitative real-time PCR are often used to measure the amount of enrichment of a particular DNA sequence by a protein-specific immunoprecipitation (1,2). Alternatively, the ChIP assay can be combined with genomic tiling micro-array (ChIP on chip) techniques, high throughput sequencing (ChIP-Seq), or cloning strategies, all of which allow for genome-wide analysis of protein-DNA interactions and histone modifications (5-8). SimpleChIP® primers have been optimized for amplification of ChIP-isolated DNA using real-time quantitative PCR and provide important positive and negative controls that can be used to confirm a successful ChIP experiment.
Explore pathways related to this product.
STRING - Known and Predicted Protein-Protein Interactions.
Except as otherwise expressly agreed in a writing signed by a legally authorized representative of CST, the following terms apply to Products provided by CST, its affiliates or its distributors. Any Customer's terms and conditions that are in addition to, or different from, those contained herein, unless separately accepted in writing by a legally authorized representative of CST, are rejected and are of no force or effect.
Products are labeled with For Research Use Only or a similar labeling statement and have not been approved, cleared, or licensed by the FDA or other regulatory foreign or domestic entity, for any purpose. Customer shall not use any Product for any diagnostic or therapeutic purpose, or otherwise in any manner that conflicts with its labeling statement. Products sold or licensed by CST are provided for Customer as the end-user and solely for research and development uses. Any use of Product for diagnostic, prophylactic or therapeutic purposes, or any purchase of Product for resale (alone or as a component) or other commercial purpose, requires a separate license from CST. Customer shall (a) not sell, license, loan, donate or otherwise transfer or make available any Product to any third party, whether alone or in combination with other materials, or use the Products to manufacture any commercial products, (b) not copy, modify, reverse engineer, decompile, disassemble or otherwise attempt to discover the underlying structure or technology of the Products, or use the Products for the purpose of developing any products or services that would compete with CST products or services, (c) not alter or remove from the Products any trademarks, trade names, logos, patent or copyright notices or markings, (d) use the Products solely in accordance with CST Product Terms of Sale and any applicable documentation, and (e) comply with any license, terms of service or similar agreement with respect to any third party products or services used by Customer in connection with the Products.