| Cat. # | Size | Qty. | Price |
|---|---|---|---|
| 4771T | 20 µl |
|
|
| 4771S | 100 µl |
|
| REACTIVITY | H M Mk |
| SENSITIVITY | Endogenous |
| MW (kDa) | 300 |
| SOURCE | Rabbit |
Product Information
For optimal ChIP results, use 10 μl of antibody and 10 μg of chromatin (approximately 4 x 106 cells) per IP. This antibody has been validated using SimpleChIP® Enzymatic Chromatin IP Kits.
| Application | Dilution |
|---|---|
| Western Blotting | 1:1000 |
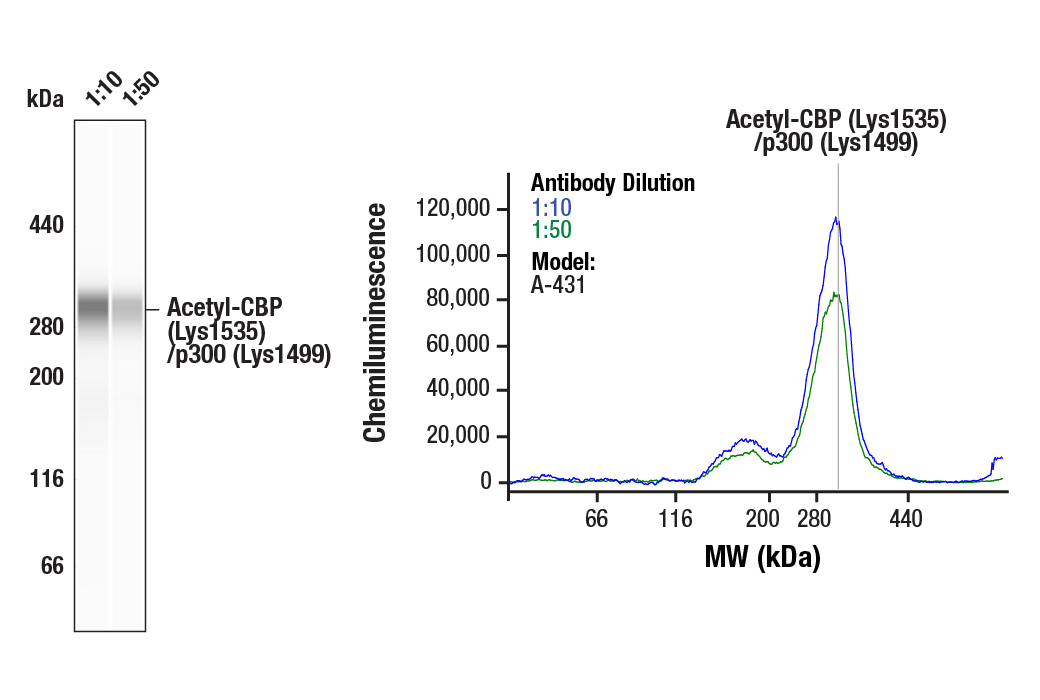
| Simple Western™ | 1:10 - 1:50 |
| Immunoprecipitation | 1:50 |
For western blots, incubate membrane with diluted primary antibody in 5% w/v BSA, 1X TBS, 0.1% Tween® 20 at 4°C with gentle shaking, overnight.
NOTE: Please refer to primary antibody product webpage for recommended antibody dilution.
From sample preparation to detection, the reagents you need for your Western Blot are now in one convenient kit: #12957 Western Blotting Application Solutions Kit
NOTE: Prepare solutions with reverse osmosis deionized (RODI) or equivalent grade water.
Load 20 µl onto SDS-PAGE gel (10 cm x 10 cm).
NOTE: Loading of prestained molecular weight markers (#59329, 10 µl/lane) to verify electrotransfer and biotinylated protein ladder (#7727, 10 µl/lane) to determine molecular weights are recommended.
NOTE: Volumes are for 10 cm x 10 cm (100 cm2) of membrane; for different sized membranes, adjust volumes accordingly.
* Avoid repeated exposure to skin.
posted June 2005
revised June 2020
Protocol Id: 10
This protocol is intended for immunoprecipitation of native proteins for analysis by western immunoblot or kinase activity utilizing Protein A magnetic separation.
NOTE: Prepare solutions with reverse osmosis deionized (RODI) or equivalent grade water.
10X Cell Lysis Buffer: (#9803) To prepare 10 ml of 1X cell lysis buffer, add 1 ml cell lysis buffer to 9 ml dH2O, mix.
NOTE: Add 1 mM PMSF (#8553) immediately prior to use.
A cell lysate pre-clearing step is highly recommended to reduce non-specific protein binding to the Protein A Magnetic beads. Pre-clear enough lysate for test samples and isotype controls.
IMPORTANT: Pre-wash #73778 magnetic beads just prior to use:
Carefully remove the buffer once the solution is clear. Add 500 μl of 1X cell lysis buffer to the magnetic bead pellet, briefly vortex to wash the beads. Place tube back in magnetic separation rack. Remove buffer once solution is clear. Repeat washing step once more.
IMPORTANT: The optimal lysate concentration will depend on the expression level of the protein of interest. A starting concentration between 250 μg/ml-1.0 mg/ml is recommended.
IMPORTANT: Appropriate isotype controls are highly recommended in order to show specific binding in your primary antibody immunoprecipitation. Use Normal Rabbit IgG #2729 for rabbit polyclonal primary antibodies, Rabbit (DA1E) mAb IgG XP® Isotype Control #3900 for rabbit monoclonal primary antibodies, Mouse (G3A1) mAb IgG1 Isotype Control #5415 for mouse monoclonal IgG1 primary antibodies, Mouse (E5Y6Q) mAb IgG2a Isotype Control #61656 for mouse monoclonal IgG2a primary antibodies, Mouse (E7Q5L) mAb IgG2b Isotype Control #53484 for mouse monoclonal IgG2b primary antibodies, and Mouse (E1D5H) mAb IgG3 Isotype Control #37988 for mouse monoclonal IgG3 primary antibodies. Isotype controls should be concentration matched and run alongside the primary antibody samples.
Proceed to one of the following specific set of steps.
NOTE: To minimize masking caused by denatured IgG heavy chains (~50 kDa), we recommend using Mouse Anti-Rabbit IgG (Light-Chain Specific) (D4W3E) mAb (#45262) or Mouse Anti-Rabbit IgG (Conformation Specific) (L27A9) mAb (#3678) (or HRP conjugate #5127). To minimize masking caused by denatured IgG light chains (~25 kDa), we recommend using Mouse Anti-Rabbit IgG (Conformation Specific) (L27A9) mAb (#3678) (or HRP conjugate #5127).
posted December 2008
revised April 2021
Protocol Id: 410
Human, Mouse, Monkey
Rat
Polyclonal antibodies are produced by immunizing animals with a synthetic acetylated peptide corresponding to residues surrounding Lys1535 of human CBP. Antibodies are purified by protein A and peptide affinity chromatography.
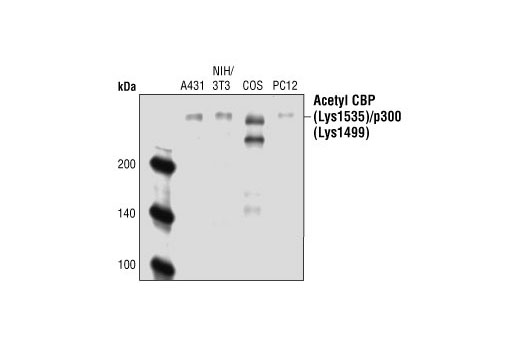
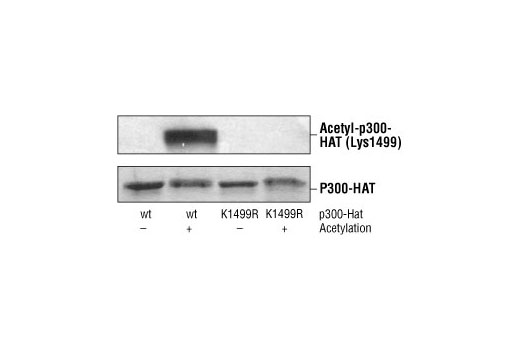
CBP (CREB-binding protein) and p300 are highly conserved and functionally related transcriptional co-activators that associate with transcriptional regulators and signaling molecules, integrating multiple signal transduction pathways with the transcriptional machinery (1,2). CBP/p300 also contain histone acetyltransferase (HAT) activity, allowing them to acetylate histones and other proteins (2). Phosphorylation of p300 at Ser89 by PKC represses its transcriptional activity, and phosphorylation at the same site by AMPK disrupts the association of p300 with nuclear receptors (3,4). Ser1834 phosphorylation of p300 by Akt disrupts its association with C/EBPβ (5). Growth factors induce phosphorylation of CBP at Ser437, which is required for CBP recruitment to the transcription complex (6). CaM kinase IV phosphorylates CBP at Ser302, which is required for CBP-dependent transcriptional activation in the CNS (7). The role of acetylation of CBP/p300 is of particular interest (2,8). Acetylation of p300 at Lys1499 has been demonstrated to enhance its HAT activity and affect a wide variety of signaling events (9).
Explore pathways related to this product.
STRING - Known and Predicted Protein-Protein Interactions.
Except as otherwise expressly agreed in a writing signed by a legally authorized representative of CST, the following terms apply to Products provided by CST, its affiliates or its distributors. Any Customer's terms and conditions that are in addition to, or different from, those contained herein, unless separately accepted in writing by a legally authorized representative of CST, are rejected and are of no force or effect.
Products are labeled with For Research Use Only or a similar labeling statement and have not been approved, cleared, or licensed by the FDA or other regulatory foreign or domestic entity, for any purpose. Customer shall not use any Product for any diagnostic or therapeutic purpose, or otherwise in any manner that conflicts with its labeling statement. Products sold or licensed by CST are provided for Customer as the end-user and solely for research and development uses. Any use of Product for diagnostic, prophylactic or therapeutic purposes, or any purchase of Product for resale (alone or as a component) or other commercial purpose, requires a separate license from CST. Customer shall (a) not sell, license, loan, donate or otherwise transfer or make available any Product to any third party, whether alone or in combination with other materials, or use the Products to manufacture any commercial products, (b) not copy, modify, reverse engineer, decompile, disassemble or otherwise attempt to discover the underlying structure or technology of the Products, or use the Products for the purpose of developing any products or services that would compete with CST products or services, (c) not alter or remove from the Products any trademarks, trade names, logos, patent or copyright notices or markings, (d) use the Products solely in accordance with CST Product Terms of Sale and any applicable documentation, and (e) comply with any license, terms of service or similar agreement with respect to any third party products or services used by Customer in connection with the Products.