According to a central dogma of biology, RNA is the molecule that translates the genomic information contained in DNA into protein. However, this simplistic view does not begin to touch upon the complexities of RNA function. The development of new reagents and techniques, such as RNA cross-linking and immunoprecipitation (CLIP) coupled with next generation sequencing, has enabled transcriptome-wide analyses that have caused research in this emerging field to explode. The result? Novel insights as to how RNA regulation contributes to the progression of a variety of diseases.
After transcription, messenger RNAs (mRNAs) interact with RNA-binding proteins (RBPs) to form messenger ribonucleoprotein (mRNP) complexes that regulate the fate of specific mRNAs (1). These include capping, splicing, transport, stability, silencing, decay, and translation (1, 2). Epitranscriptomics, which refers to post-transcriptional modifications of RNA, including adenosine and cytosine methylation, adenosine-to-inosine editing, and pseudouridylation, also play a key role in influencing mRNA fate and adds another layer of complexity to the regulation of gene expression.
N6-methyladenosine (m6A) is the most prevalent and well understood RNA modification, and, akin to histone modifications, there are m6A-specific writers, readers, and erasers. m6A is catalyzed by the METTL3/METTL4 methyltransferase complex to enable interactions with m6A-binding proteins, such as YTHDC, YTHDF, and eIF3. These interactions mediate m6A-dependent cellular events like the promotion of translation and RNA stability. m6A modifications are reversible by demethylases like FTO and ALKBH5. Evidence indicates m6A modifications play a role in development, T-cell homeostasis, and the pluripotency of embryonic stem cells (3).
Aberrant m6A modifications are implicated in a variety of cancers (4). The mechanisms involved, where writers, readers, and erasers promote tumorigenesis, appear to be specific to the target mRNA affected and the cancer type. For example, increased levels of METTL3 in acute myeloid leukemia (AML) leads to the overexpression of anti-apoptotic oncogene BCL-2, while decreased levels of METTL3 in glioblastomas leads to increased expression of the oncogene ADAM19.
Evidence suggests m6A modifications also play a role in dopaminergic neuronal death, a hallmark of Parkinson’s disease (PD) (5). Decreased levels of global m6A are observed in the striatum of PD rat brains, and the overexpression of FTO in dopaminergic cells induces the expression of the NMDA receptor 1 and elevates oxidative stress and Ca2+ levels to induce apoptosis. Genetic variants in intron 1 and intron 2 of the FTO gene has also been associated with Alzheimer’s disease (AD). SNPs in these regions were observed in Caucasians of European ancestry and Caribbean Hispanics. FTO was also expressed at lower levels in AD patients compared to control (6).
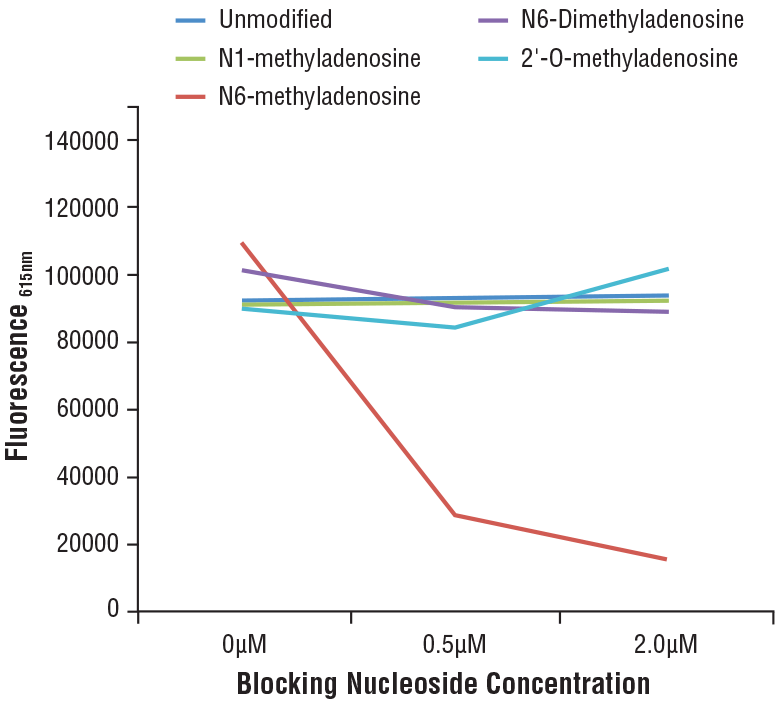
N6-Methyladenosine (m6A) (D9D9W) Rabbit mAb #56593: Specificity of N6-Methyladenosine (m6A) (D9D9W) Rabbit mAb was determined by competitive ELISA. The graph depicts the binding of the antibody to a pre-coated m6A oligonucleotide in the presence of increasing concentrations of differentially modified adenosine. As shown in the graph, antibody binding is only blocked by free m6A nucleoside.
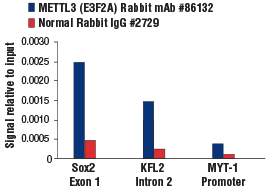
METTL3 (E3F2A) Rabbit mAb #86132: Chromatin immunoprecipitations were performed with crosslinked chromatin from mES cells and either METTL3 (E3F2A) Rabbit mAb or Normal Rabbit IgG #2729 using SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) #9005. The enriched DNA was quantified by real-time PCR using SimpleChIP® Mouse Sox2 Exon1 Primers #30180, mouse KLF2 intron 2 primers, and SimpleChIP® Mouse MYT-1 Promoter Primers #8985. The amount of immunoprecipitated DNA in each sample is represented as signal relative to the total amount of input chromatin, which is equivalent to one.
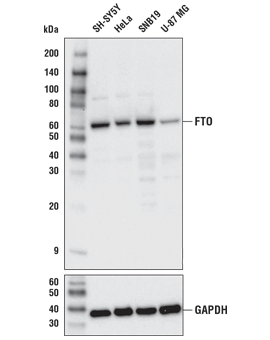
FTO (D6Z8W) Rabbit mAb #31687: Western blot analysis of extracts from various cell lines using FTO (D6Z8W) Rabbit mAb (upper) or GAPDH (D16H11) XP® Rabbit mAb #5174 (lower).
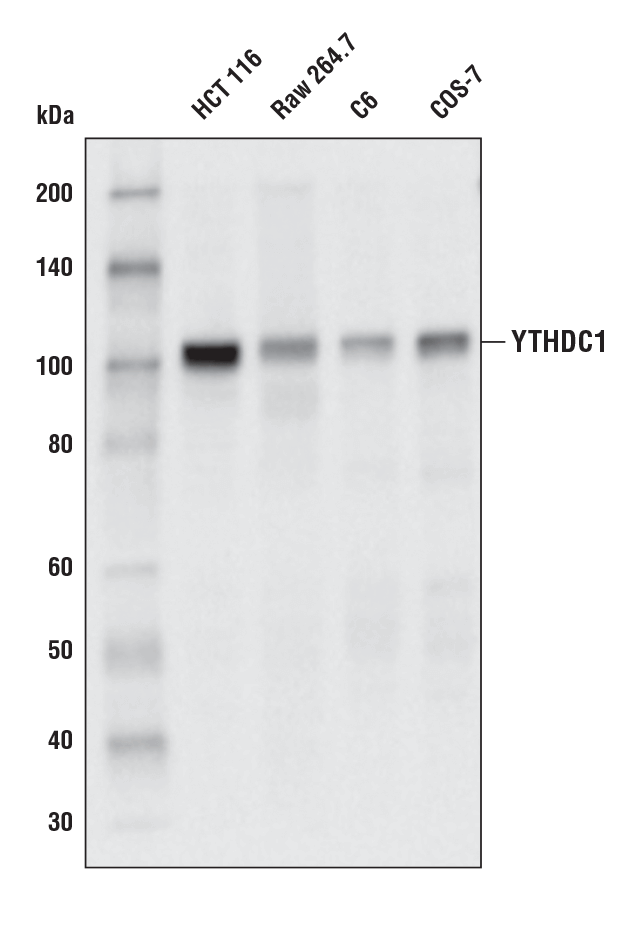
YTHDC1 (E4I9E) Rabbit mAb #77422: Western blot analysis of extracts from various cell lines using YTHDC1 (E4I9E) Rabbit mAb.
mRNAs can contain an N7-methylguanosine (m7G) modification at the 5’ end that is responsible for the recruitment of initiation factors and gives cells the ability to discriminate between self and viral mRNA (7). A reversible methyl modification occurs when the first nucleotide after the m7G cap, 2′-O-methyladenosine (Am), is further methylated by the writer CAPAM/PCIF1 to form N6,2′-O-dimethyladenosine (m6Am) (7). This methyl group can then be removed by the m6Am eraser FTO. Studies suggest methylation by CAPAM/PCIF1 promotes translation of the capped mRNA, while other studies observed increased stability in the m6Am modified RNA compared to its Am counterpart.
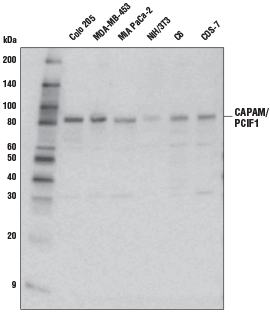
CAPAM/PCIF1 Antibody #98085: Western blot analysis of extracts from various cell lines using CAPAM/PCIF1 Antibody.
Unlike m6A and m6Am, the N1-methyladenosine (m1A) modification perturbs Watson-Crick base pairing to potentially interfere with protein-RNA interactions (8). It is also thought to regulate tRNA and rRNA stability. There is significantly less known about this modification compared to the m6A and m6Am modifications due to challenges identifying it. The presence of m1A modifications in mRNA is inconclusive, potentially due to differences in the sequencing technologies used, as some groups have detected it in abundance while other groups have not detected it at all. Groups that have observed m1A modifications in mRNA have identified ALKBH3 as an mRNA m1A eraser and have yet to identify an m1A writer (9).
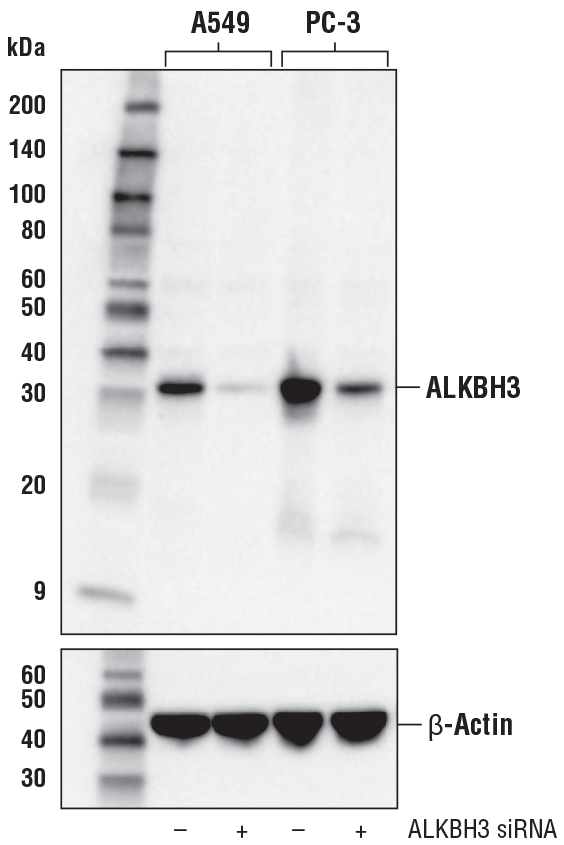
ALKBH3 (E6S4R) Rabbit mAb #87620: Western blot analysis of extracts from A549 and PC-3 cells, mock transfected (-) or transfected with ALKBH3 siRNA (+), using ALKBH3 (E6S4R) Rabbit mAb (upper) or β-Actin (D6A8) Rabbit mAb #8457 (lower).
Pseudouridine is a modification where the nitrogen-carbon bond in uracil is replaced with a carbon-carbon bond. Pseudouridine synthases (PUS proteins), such as Dyskerin, catalyze this change primarily on tRNAs to promote tRNA stability (10). PUS proteins also modify rRNAs to assist in ribosome assembly, which is required for proper protein synthesis. mRNA pseudouridylation has only recently been observed and may influence mRNA splicing, translation, half-life, and translational regulation.
Abnormal activity of the Dyskerin protein correlates with cancer progression and poor patient prognosis. Pseudouridylation also plays a role in regulating viral latency processes in HIV infections and is implicated in Crohn’s disease, celiac disease, X-linked dyskeratosis congenita (X-DC), maternally inherited diabetes and deafness (MIDD), and mitochondrial myopathy and sideroblastic anemia (MLASA).
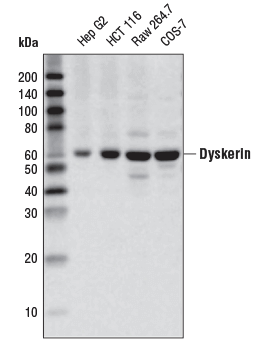
Dyskerin (D6N4K) Rabbit mAb #53234: Western blot analysis of extracts from various cell lines using Dyskerin (D6N4K) Rabbit mAb.
Adenosine deaminases acting on RNAs (ADARs) catalyze the deamination of adenosine to form inosine in pre-mRNAs. This reaction, also known as A-to-I editing, alters RNA splicing and RNA maturation and can result in changes to protein coding and function. In addition, recent studies have shown that A-to-I editing prevents the innate immune response against self RNA (11). Editing by ADAR1 can destabilize double-stranded RNAs (dsRNAs) and suppress the activation of RIG-I or MDA5, two surveillance proteins involved in the recognition and clearance of nonself materials.
A-to-I editing contributes to cancer progression by altering the coding and behavior of proteins like AZIN1. A-to-I editing of AZIN1 results in a serine-to-glycine substitution at residue 367, which enhances its ability to suppress the degradation of multiple oncoproteins that then act to promote cell motility, invasion, and metastasis. In addition, the loss or knockdown of ADAR1 in cancer cells overcomes resistance to cancer immunotherapies (12).
A-to-I editing appears to modulate HIV infection; however, there is not yet a consensus on how it regulates the disease. Although A-to-I editing of HIV-1 RNA has been shown to disrupt viral assembly and inhibit propagation in primary macrophages, increased ADAR1 activity correlates with increased HIV-1 viral replication in CD4+ T-cells (11). More work is required to understand the effect of A-to-I editing on HIV infection.
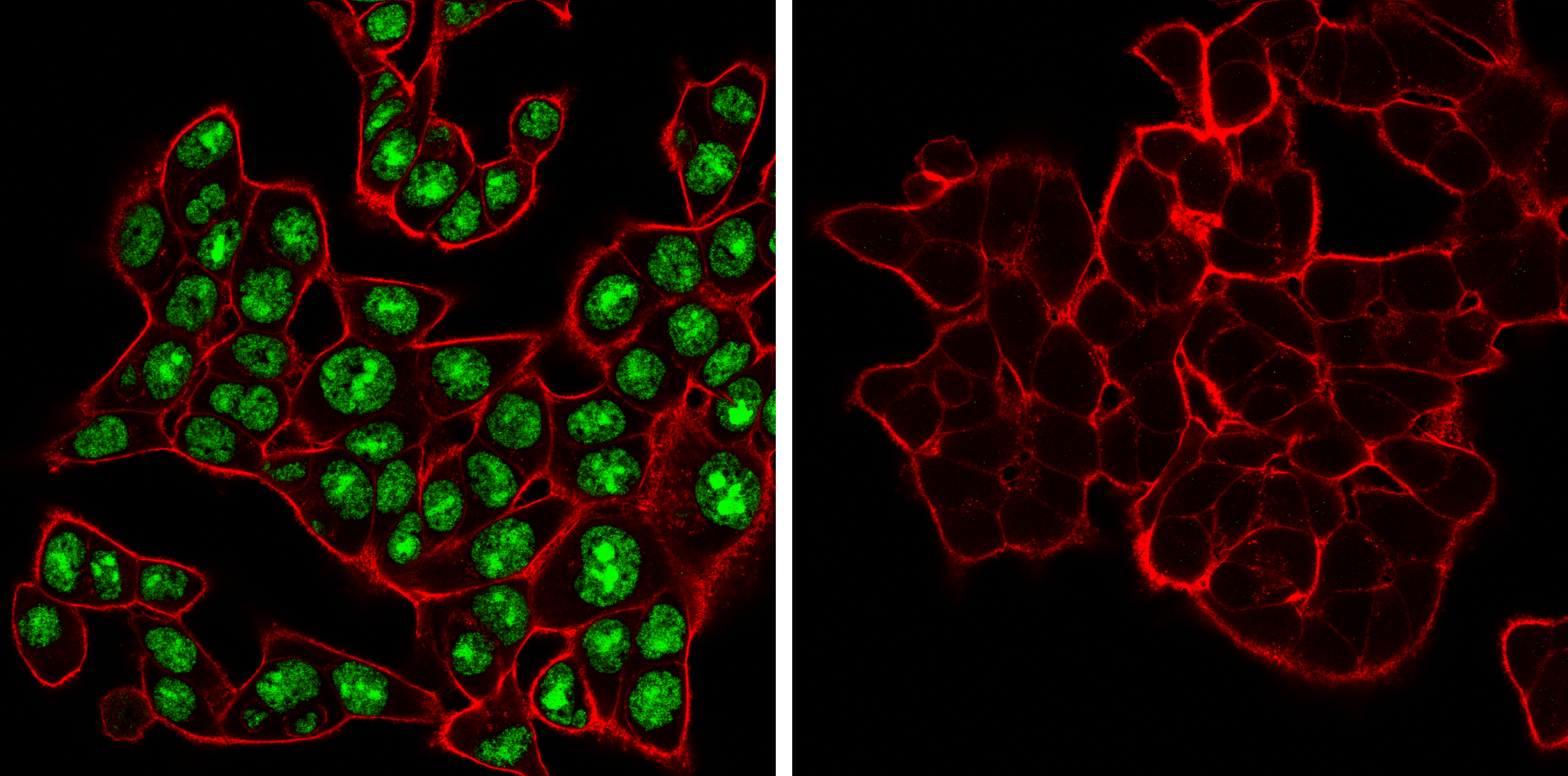
ADAR (E6X9R) XP® Rabbit mAb #81284: Confocal immunofluorescent analysis of HCT 116 cells, either wild-type (left, positive) or ADAR1 knockout (right, negative), using ADAR1 (E6X9R) XP® Rabbit mAb (green). Actin filaments were labeled with DyLight 650 Phalloidin #12956 (red).
Alternative splicing is a regulated process where alternative mRNA transcripts are generated from a single gene, resulting in the translation of different isoforms of the same protein which can confer distinct functionality, binding partners, and/or subcellular localization. The basic modes of alternative splicing include constitutive splicing, intron retention, exon skipping, alternative splice site selection, mutually exclusive splicing, alternative promoter selection, and alternative polyadenylation sites (13). Alternative splicing is regulated by proteins such as PTBP1 and heterogeneous nuclear ribonucleoproteins like hnRNP A1 bound to pre-mRNA and catalyzed by spliceosomes
Spliceosomes are found within the nucleus of eukaryotic cells and are large molecular machines assembled from small nuclear RNAs (snRNAs) and proteins. Major spliceosomes are the most abundant in eukaryotic cells and contain snRNPs with U1, U2, U4, U5, and U6 snRNAs, while minor spliceosomes are much less common and contain snRNPs with U11, U12, U4atac, U4, and U5 snRNAs (14). Spliceosomes recognize different sequence elements, such as a 5’ end splice site, in pre-mRNA before removing noncoding introns and stitching the flanking exons back together to create the mature spliced mRNA. The core spliceosome also recruits proteins, such as SAM68, to assist with alternative splicing.
SAM68 is an RBP that regulates the alternative splicing of CD44 mRNA and other RNAs. Many activities of SAM68 and its alternative splicing targets are associated with the development of spinal muscular atrophy and cancers (15). TDP-43, a protein that regulates RNA splicing, stability, and transport, is abnormally ubiquitinated, phosphorylated, and cleaved to generate fragments that form insoluble aggregates in patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis (16). Finally, UAP56, an ATP-dependent splicing factor, also promotes the androgen receptor AR-V7 splice variant associated with metastatic castration resistant prostate cancer (17, 18). Other splice variants known to play a key role in the progression of cancer and/or resistance to treatment include the 12 different p53 splice variants linked to a variety of cancers (19). Clinical studies indicate that an expression profile of p53 isoforms can be linked with tumor progression and clinical response.
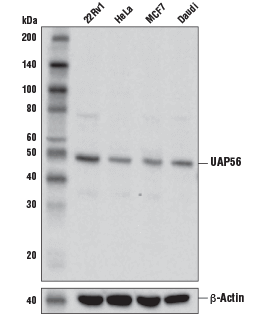
UAP56 Antibody #55509: Western blot analysis of extracts from various cell lines using UAP56 Antibody (upper) and β-Actin (D6A8) Rabbit mAb #8457 (lower).
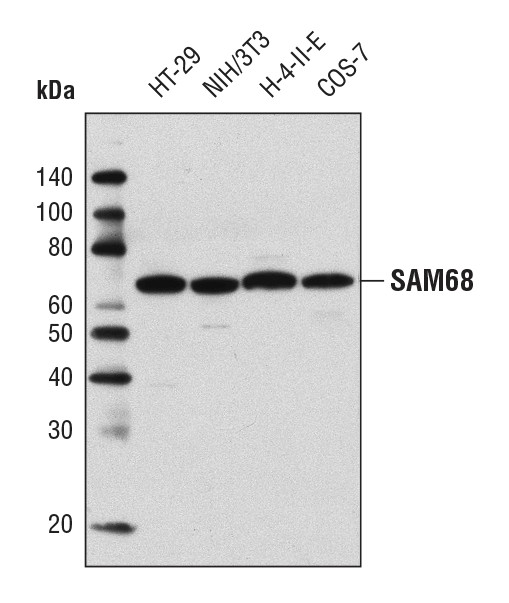
SAM68 Antibody #12538: Western blot analysis of extracts from various cell lines using SAM68 Antibody.
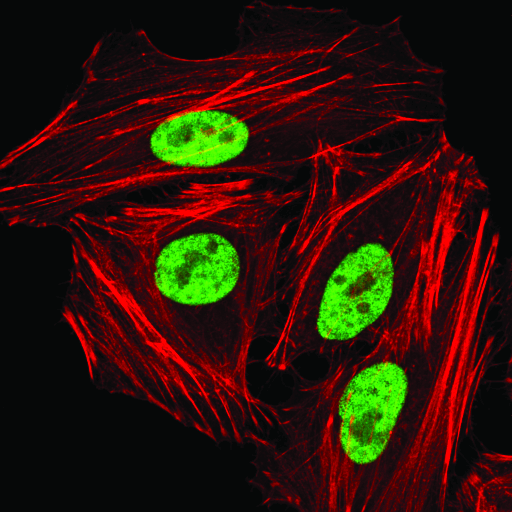
hnRNP A1 (D21H11) Rabbit mAb #8443: Confocal immunofluorescent analysis of HeLa cells using hnRNP A1 (D21H1) Rabbit mAb (green). Actin filaments were labeled with DyLight 554 Phalloidin #13054 (red).
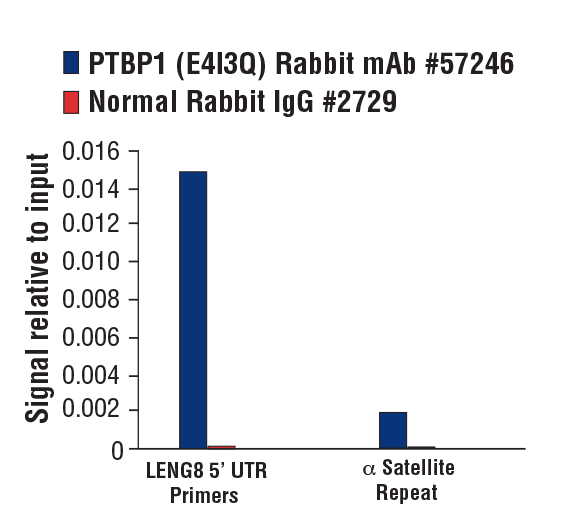
PTBP1 (E4I3Q) Rabbit mAb #57246: Chromatin immunoprecipitations were performed with cross-linked chromatin from MCF7 cells grown in phenol red free medium and 5% charcoal stripped FBS for 4 d followed by treatment with β-estradiol (10 nM, 45 min) and either PTBP1 (E4I3Q) Rabbit mAb or Normal Rabbit IgG #2729 using SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) #9005. The enriched DNA was quantified by real-time PCR using SimpleChIP® Human LENG8 5’ UTR Primers #70823 and SimpleChIP® Human α Satellite Repeat Primers #4486. The amount of immunoprecipitated DNA in each sample is represented as signal relative to the total amount of input chromatin, which is equivalent to one.
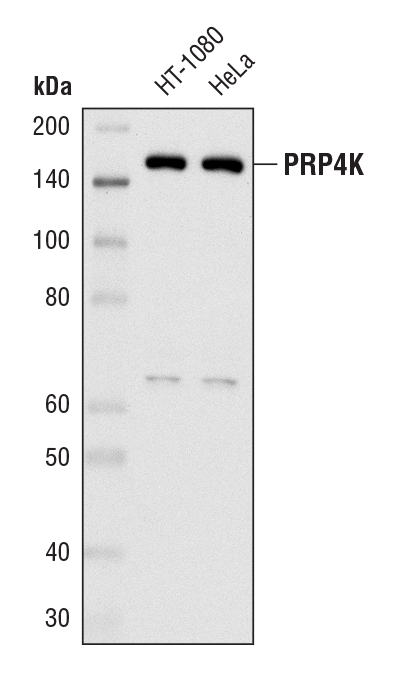
PRP4K (D27A1) Rabbit mAb #8577: Western blot analysis of extracts from HT-1080 and HeLa cells using PRP4K (D27A1) Rabbit mAb.
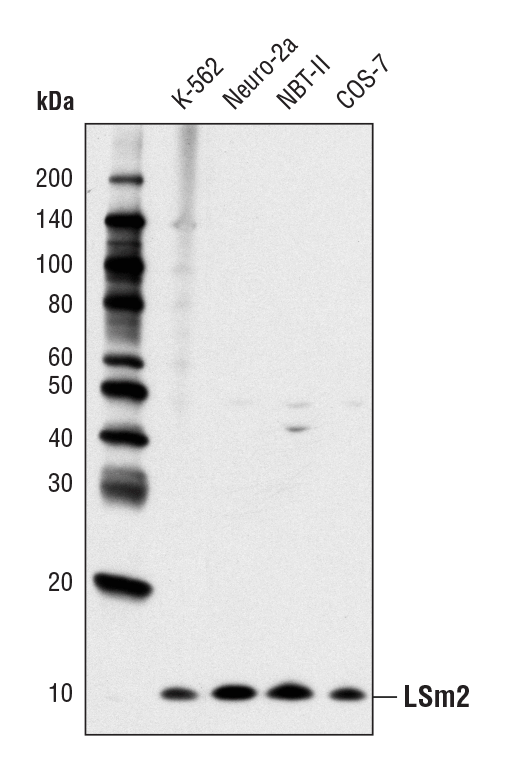
LSm2 (D9X6C) Rabbit mAb #13119: Western blot analysis of extracts from various cell lines using LSm2 (D9X6C) Rabbit mAb.
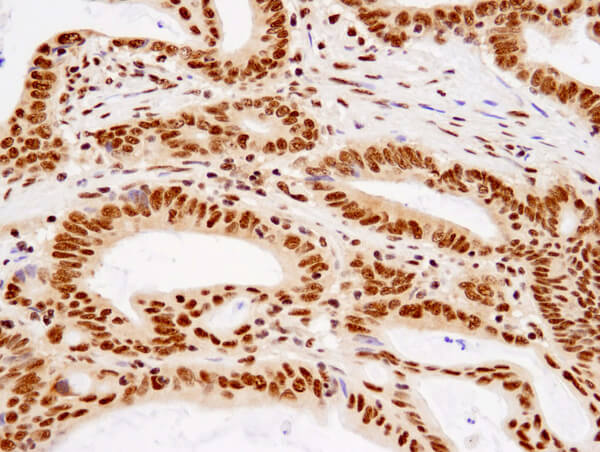
TDP43 (D9R3L) Rabbit mAb #89789: Immunohistochemical analysis of paraffin-embedded human colon carcinoma using TDP43 (D9R3L) Rabbit mAb
miRNAs are noncoding RNAs that either degrade or inhibit expression of target mRNAs and regulate post-transcriptional gene expression (20). They are transcribed by RNA polymerase II into primary miRNAs (pri-mRNAs), which are then cleaved by Drosha and DGCR8 in the nucleus to form pre-miRNAs. These pre-miRNAs, which contain short hairpins, are subsequently exported to the cytoplasm by XPO5 where they are further processed by Dicer to become mature miRNAs. The mature miRNA then interacts with proteins like Argonaute 2 (AGO2) and TRBP to form the RNA-induced silencing complex (RISC). RISC-dependent gene silencing occurs either through mRNA degradation or translation inhibition.
Perturbation in the miRNA machinery, such as altered expression of AGO2, Drosha, and DGCR8, have been shown to promote carcinogenesis in many disease contexts. miRNA dysregulation has also been linked to heart and kidney disease (21, 22).
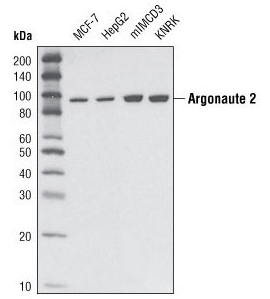
Argonaute 2 (C34C6) Rabbit mAb #2897: Western blot analysis of extracts from various cell types using Argonaute 2 (C34C6) Rabbit mAb.
TRBP2 (D7C8K) Rabbit mAb #62043: Western blot analysis of extracts from various cell lines using TRBP2 (D7C8K) Rabbit mAb.
Drosha (D28B1) Rabbit mAb #3364: Western blot analysis of extracts from various cell types using Drosha (D 28B1) Rabbit mAb.

Dicer (D38E7) Rabbit mAb #5362: Western blot analysis of extracts from MCF7 and 293 cells using Dicer (D38E7) Rabbit mAb.
DGCR8 (D78E4) Rabbit mAb #6914: Western blot analysis of extracts from 293 and HeLa cells using DGCR8 (D78E4) Rabbit mAb.
mRNAs interact with RBPs to form mRNP granules that regulate mRNA location, stability, and translation. Of the many different types of granules that exist, stress granules and processing bodies are the best characterized (23). Processing bodies are cytoplasmic RNA granules that contain decapping enzymes, translational regulators, miRNA machinery, and factors associated with nonsense-mediated decay. Stress granules safeguard specific mRNAs during cellular stress and contain elongation factors, 40 ribosomal subunits, and stalled mRNAs. They form in response to stress-induced phosphorylation of eIF2. G3BP1, a protein that interacts with the SH3 domain of RasGAP, is often used as a stress granule marker, as it is essential for the assembly of stress granules and functions as a stress granule nucleating protein (24).
The abnormal or persistent formation of stress granules may contribute to degenerative diseases like amyotropic lateral sclerosis, Paget’s diseases, and frontotemporal lobar degeneration (25). Cells afflicted with these disease states contain inclusion bodies that contain many of the same components as stress granules. Additionally, many mutated proteins implicated in these diseases are either found in stress granules or drive abnormal stress granule formation. Evidence also suggests a relationship between stress granules and cancer; however, the mechanisms involved to drive disease progression have yet to be elucidated (23).
XRN1 Antibody #70205: Western blot analysis of extracts from KARPAS-299, HDLM-2, and U-2 OS cell lines using XRN1 Antibody (upper) and α-Actinin (D6F6) XP® Rabbit mAb #6487 (lower). KARPAS cell line source: Dr. Abraham Karpas at the University of Cambridge.
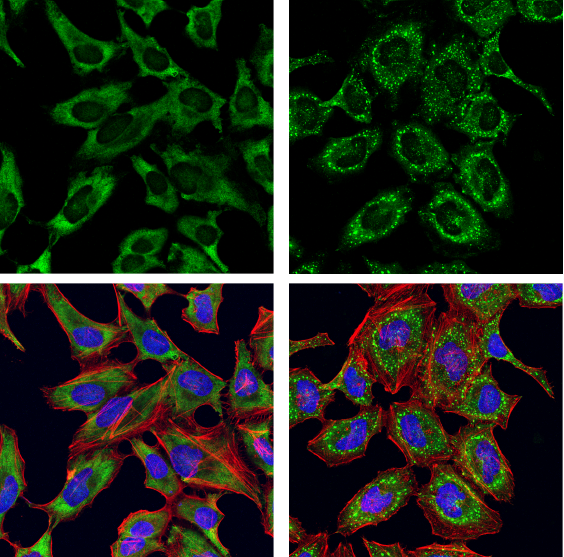
G3BP1 Antibody #17798: Confocal immunofluorescent analysis of HeLa cells, serum-starved (left) or serum-starved and then treated with sodium arsenite (500 μM, 30 min; right), using G3BP1 Antibody (green). Actin filaments were labeled with DyLight 554 Phalloidin #13054 (red). Samples were mounted in ProLong Gold Antifade Reagent with DAPI #8961 (blue).
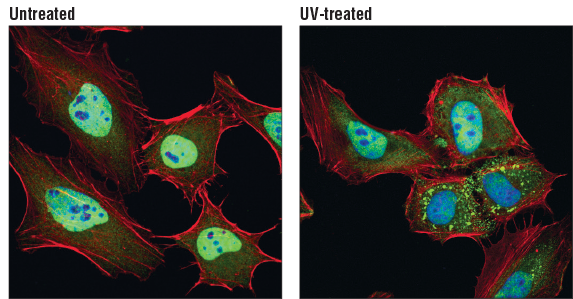
TIAR (D32D3) XP® Rabbit mAb #8509: Confocal immunofluorescent analysis of HeLa cells, untreated (left) or UV-treated (right), using TIAR (D32D3) XP® Rabbit mAb (green). Actin filaments were labeled with DyLight 554 Phalloidin #13054 (red). Blue pseudocolor = DRAQ5 #4084 (fluorescent DNA dye)
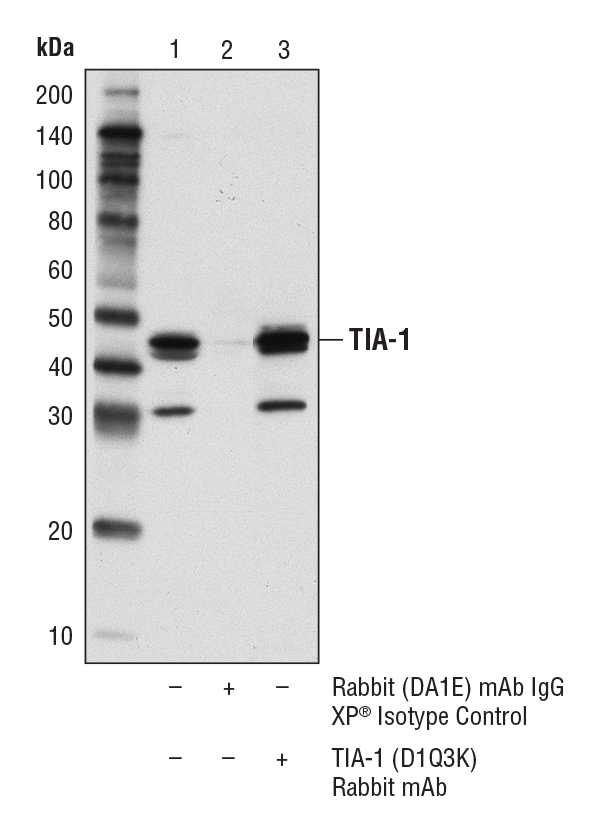
TIA-1 (D1Q3K) Rabbit mAb #86050: Immunoprecipitation of TIA-1 from KARPAS-299 cell lysates. Lane 1 represents 10% input, lane 2 is Rabbit (DA1E) mAb IgG XP® Isotype Control #3900, and lane 3 is TIA-1 (D1Q3K) Rabbit mAb. Western blot was performed using TIA-1 (D1Q3K) Rabbit mAb. A conformation-specific secondary antibody was used to avoid cross-reactivity with IgG. Cell line source: Dr. Abraham Karpas at the University of Cambridge.