An enzyme-linked immunosorbent assay (ELISA) is a specific type of enzyme immunoassay (EIA) that allows for the quantitation of a molecule of interest using antibodies. Antibodies are used to specifically identify the analyte (eg, peptides, proteins, antibodies, small molecules). An enzyme, such as horseradish peroxidase (HRP), is either directly or indirectly coupled to the antibody in order to provide the detection method and possible signal amplification.
An ELISA differs from other immunoassays in that this technique amplifies signals while using a sample in solution.
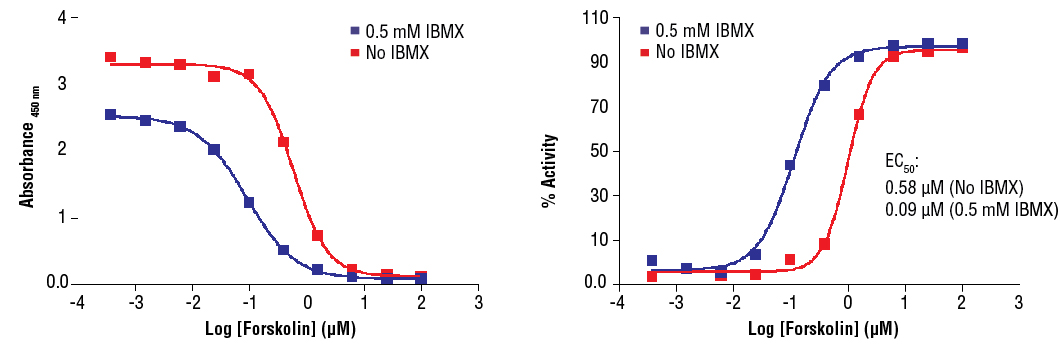
Cyclic AMP XP® Assay Kit #4339: Treatment of CHO cells with Forskolin #3828 increases cAMP concentration as detected by Cyclic AMP XP® Assay Kit #4339. CHO cells were seeded at 4x104 cells/well in a 96-well plate and incubated overnight. Cells were either left untreated or pretreated with 0.5 mM IBMX for 30 minutes prior to forskolin treatment (15 minutes) and lysed with 1X Cell Lysis Buffer #9803. The absorbance values (left) and percentage of activity (right) are shown above. The percentage of activity is calculated as follows: % activity=100x[(A-Abasal)/(Amax-Abasal)], where A is the sample absorbance, Amax is the absorbance at maximum stimulation (i.e., high forskolin concentration), and Abasal is the absorbance at basal level (no forskolin). Forskolin directly activates adenylyl cyclases and increases cellular cAMP concentration. IBMX is a non-specific inhibitor of cAMP and cGMP phosphodiesterases and promotes accumulation of cAMP and cGMP in cells.
ELISAs deliver a simple, robust, and cost-effective method to analyze and quantify one or more antigens from a variety of sample types, such as cell lysate, tissue lysate, or serum.
ELISA is a popular technique for research and diagnostic samples and can be used as both a single sample test or high throughput method for screening.
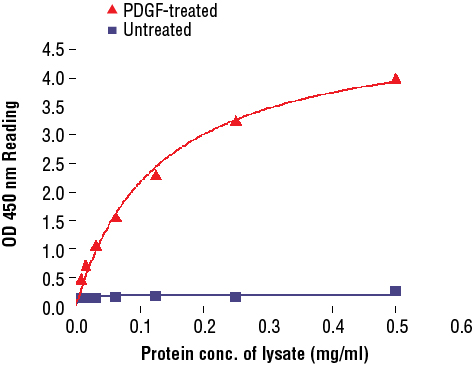
PathScan® Phospho-Akt (Thr308) Sandwich ELISA Kit #7252: The relationship between protein concentration of lysates from untreated and PDGF-treated NIH/3T3 cells and kit assay optical density readings is shown. After starvation, NIH/3T3 cells (85% confluence) were treated with PDGF (50 ng/ml) for 10 min at 37°C, and then lysed.
There are several experimental set-ups depending on the type of ELISA carried out.
All these set-ups are typically performed in a micro-well plate to which either the antigen or capture antibody is adsorbed. They rely on the amplification of signal from the antibody enzyme conjugate, which will bind to the antigen of interest.
The added enzymatic substrate will produce either a change in color, fluorescence, or luminescence and be detected and then quantified.
The success of the experiment and reliability of the results are predicated upon highly specific binding of the chosen antibodies to the antigen of interest. Along with specificity, the chosen antibodies should have high affinity and avidity for the antigen.
ELISA is considered a gold standard for quantitative analysis of biologic samples due to the specificity of antibody reagents and simplicity of the assay.
ELISAs can be broadly used in multiple research areas as well as diagnostic screening. Cell Signaling Technology offers ELISA solutions for research areas such as:
ELISA cannot be used when spatial resolution is required to analyze results.
In contrast to western blots, ELISA provides a more accurate quantitation and considers molecules in their nondenatured form.
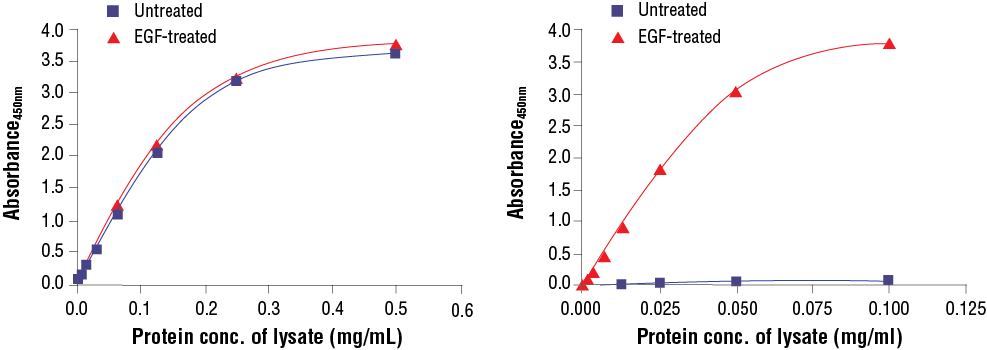
FastScan™ Total compared to FastScan™ Phospho: Treatment of A-431 cells with EGF stimulates phosphorylation of p44/42 MAPK (Erk1/2) at Thr202 and Tyr204, but does not affect the level of total p44/42 MAPK (Erk1/2). The relationship between lysate protein concentration from untreated and EGF-treated A-431 cells and the absorbance at 450 nm using the FastScan™ Total p44/42 MAPK (Erk1/2) ELISA Kit #67404 is shown in the left figure.
Treatment of A-431 cells with EGF stimulates phosphorylation of p44/42 MAPK (Erk1/2) at Thr202 and Tyr204 but does not effect the level of total p44/42 MAPK (Erk1/2). The relationship between lysate protein concentration from untreated and EGF-treated A-431 cells and the absorbance at 450 nm using the FastScan™ Phospho-p44/42 MAPK (Erk 1/2) (Thr202/Tyr204) ELISA Kit #42173 is shown in the right figure.