| Cat. # | Size | Qty. | Price |
|---|---|---|---|
| 56795S | 1 Kit (24 assays) |
|
| Product Includes | Volume (with Count) | Storage Temp | |||
|---|---|---|---|---|---|
| End Prep Enzyme Mix | 1 x 72 µl | -20°C | |||
| End Prep Reaction Buffer | 1 x 168 µl | -20°C | |||
| Ligation Enhancer | 1 x 24 µl | -20°C | |||
| Ligation Master Mix | 1 x 720 µl | -20°C | |||
| Q5 PCR Master Mix | 1 x 600 µl | -20°C |
Product Information
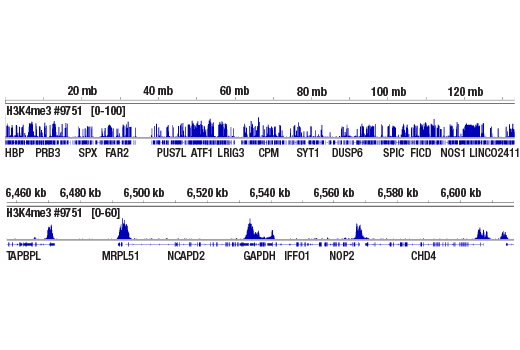
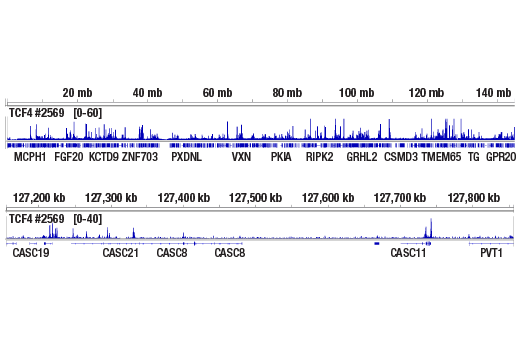
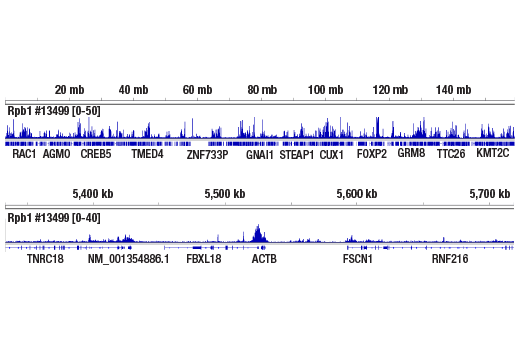
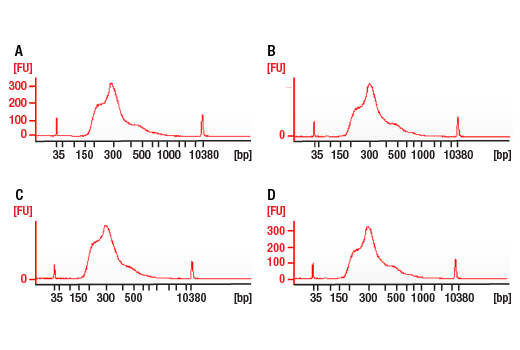
Next generation sequencing (NG-seq) is a high throughput method that can be used downstream of chromatin immunoprecipitation (ChIP) and Cleavage Under Targets and Release Using Nuclease (CUT&RUN) assays to identify and quantify target DNA enrichment across the entire genome. The DNA Library Prep Kit for Illumina Systems (ChIP-seq, CUT&RUN) contains all of the enzymes and buffers necessary to generate high quality DNA sequencing libraries from ChIP DNA or CUT&RUN DNA for next-generation sequencing on the Illumina Systems platform. The fast, user-friendly workflow minimizes hands-on time needed for generation and purification of DNA libraries.
Each kit component must pass rigorous quality control standards, and for each new lot the entire set of reagents is functionally validated together by construction and sequencing of indexed libraries on the Illumina Systems sequencing platform.
This product must be used in combination with Multiplex Oligos for Illumina Systems (Single Index Primers) (ChIP-seq, CUT&RUN) #29580 or Multiplex Oligos for Illumina Systems (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538. This product provides sufficient amounts of reagents for 24 reactions and is compatible with both enzymatic- or sonication-fragmented, ChIP-enriched DNA, and CUT&RUN DNA (with different protocols for DNA enriched from ChIP or CUT&RUN assays).
Compatible Assay kits:
SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) #9003
SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) #9005
SimpleChIP® Plus Sonication Chromatin IP Kit #56383
Multiplex Oligos for Illumina Systems (Single Index Primers) (ChIP-seq, CUT&RUN) #29580
Multiplex Oligos for Illumina Systems (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538
CUT&RUN Assay Kit #86652
Non-Compatible SimpleChIP® kits:
SimpleChIP® Enzymatic Chromatin IP Kit (Agarose Beads) #9002
SimpleChIP® Plus Enzymatic Chromatin IP Kit (Agarose Beads) #9004
Note: Agarose beads are blocked with sonicated salmon sperm DNA, which will contaminate DNA
library preps and NG-seq.
Required Reagents
Reagents Included:
Reagents Not Included:
| SAFE STOP | This is a safe stopping point in the protocol, if stopping is necessary. |
When performing ChIP-seq, it is necessary to generate a control DNA sequencing library that can be used to
determine any experimental bias in DNA enrichment that is introduced during the ChIP assay and DNA library
preparation. DNA purified from input chromatin (i.e. the chromatin that was used for the IP) is typically
used to generate the control DNA library.
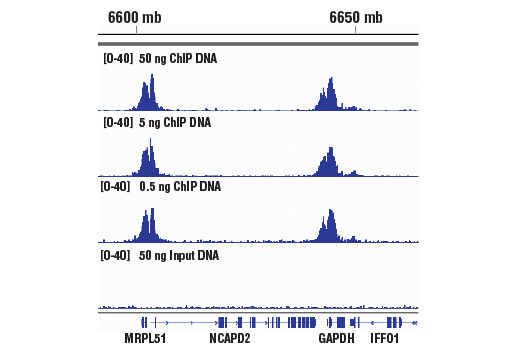
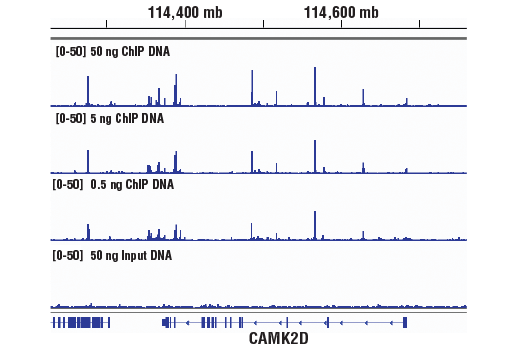
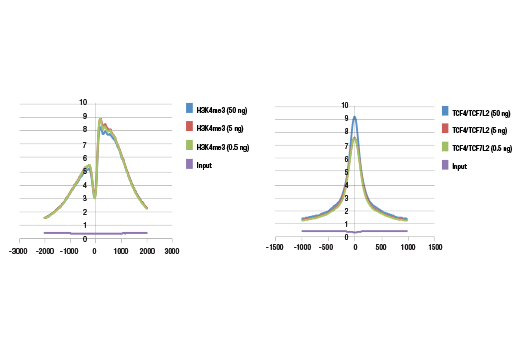
Starting Material: 500 pg –1 μg of ChIP DNA. To ensure optimal diversity of the DNA
sequencing libraries, we recommend using 5 ng of ChIP-enriched DNA for transcription factor or co-factor
ChIP-seq, 50 ng of ChIP-enriched DNA for total histone or histone modification ChIP-seq, and 50 ng of input
DNA for the control DNA sequencing library. If necessary, less than 5 ng of ChIP-enriched DNA can be used
for library generation; however, this may result in a lower diversity of library due to PCR bias during
amplification.
Before starting:
Before starting:
Size selection is NOT recommended during the Cleanup Adaptor-ligated ChIP DNA phase, because it results in a dramatic decrease in both yield and diversity of ChIP-seq DNA libraries.
Before starting:
Before starting:
| Reagents | Volume for 1 PCR Reaction (50 μl) |
| Purified adaptor ligated ChIP-DNA fragments (from step 9, Section III) | 15 μl |
| Q5 PCR Master Mix (•) | 25 μl |
| Single Index Primer for Illumina Systems (•) (or Dual Index 7 Primer for Illumina Systems [orange cap] for dual indexing) | 5 μl |
| Universal PCR Primer for Illumina Systems (•) (or Dual Index 5 Primer for Illumina Systems [white cap] for dual indexing) | 5 μl |
| a. | Initial Denaturation | 98°C 30 sec |
| b. | Denaturation | 98°C 10 sec |
| c. | Anneal and Extension | 65°C 15 sec |
| d. |
For starting material of 50 ng ChIP DNA, repeat steps b and c for a total of 6
cycles.
|
|
| e. | Final Extension | 65°C 3 min |
| f. | Hold | 4°C |
Before starting:
| SAFE STOP | This is a safe stopping point in the protocol, if stopping is necessary. |
When performing CUT&RUN-seq, it is necessary to generate a control DNA sequencing library that can be
used to determine any experimental bias in DNA enrichment that is introduced during the CUT&RUN assay
and DNA library preparation. DNA purified from input DNA (refer to Section V of CUT&RUN Assay Kit #86652
Protocol) is typically used to generate the control DNA library.
Starting Material: 0.5 ng–1 µg of CUT&RUN DNA. The typical yield for CUT&RUN DNA is
0.6 to 6 ng per reaction with 100,000 starting cells. To ensure optimal diversity of the DNA sequencing
libraries, we recommend using as much CUT&RUN DNA as you can obtain from a CUT&RUN reaction. If
necessary, as little as 0.1 ng of CUT&RUN DNA can be used for library generation; however, this may
result in a lower diversity of library due to PCR bias during amplification. For the control DNA sequencing
library using input DNA, 5 - 10 ng is a good starting point. Please note that a Picogreen assay is needed to
determine the concentration of CUT&RUN DNA because Nanodrop alone is not sensitive enough.
Before starting:
Before starting:
| Starting DNA | Adaptor Dilution | Working Adaptor Concentration |
| 100 ng – 1 μg | No Dilution | 15 μM |
| 5 ng – 100 ng | 1:10 | 1.5 μM |
| 2.5 ng – 5 ng | 1:25 | 0.6 μM |
| 1.25 ng – 2.5 ng | 1:50 | 0.3 μM |
| Less than 1.25 ng | 1:125 | 0.12 μM |
Size selection is NOT recommended during the Cleanup Adaptor-ligated CUT&RUN DNA phase, because it results in a dramatic decrease in both yield and diversity of DNA libraries.
Before starting:
Before starting:
| Reagents | Volume for 1 PCR Reaction (50 μl) |
| Purified adaptor ligated CUT&RUN-DNA fragments (from step 9, Section III) | 15 μl |
| Q5 PCR Master Mix ( •) | 25 μl |
| Single Index Primer for Illumina Systems ( •) (or Dual Index 7 Primer for Illumina Systems [orange cap] for dual indexing) | 5 μl |
| Universal PCR Primer for Illumina Systems (•) (or Dual Index 5 Primer for Illumina Systems [white cap] for dual indexing) | 5 μl |
| a. | Initial Denaturation | 98°C for 30 sec |
| b. | Denaturation | 98°C for 10 sec |
| c. | Anneal and Extension | 65°C for 13 sec |
| d. | For 50 - 100 ng starting CUT&RUN DNA | Repeat steps b and c for a total of 6 - 7 cycles |
| For 5 - 50 ng starting CUT&RUN DNA | Repeat steps b and c for a total of 8-12 cycles | |
| For 1 - 5 ng starting CUT&RUN DNA | Repeat steps b and c for a total of 13 - 15 cycles | |
| For 0.5 - 1 ng starting CUT&RUN DNA | Repeat steps b and c for a total of 16 - 17 cycles | |
| For 0.2 – 0.5 ng starting CUT&RUN DNA | Repeat steps b and c for a total of 18 - 19 cycles | |
| For 0.1 -0.2 ng starting CUT&RUN DNA | Repeat steps b and c for a total of 20 cycles | |
| e. | Final Extension | 65°C for 3 min |
| f. | Hold | 4°C |
Before starting:
Description
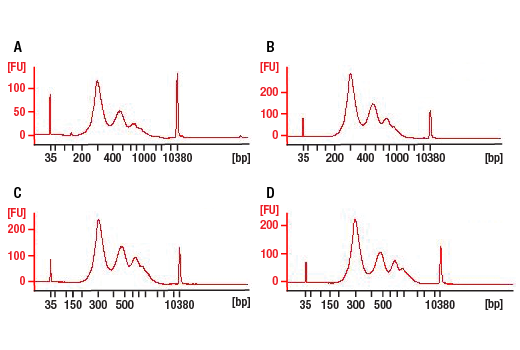
End Prep Enzyme Mix is optimized to convert 500 pg-1 μg of fragmented DNA to repaired DNA having
5´-phosphorylated, 3´-dA-tailed ends.
Quality Control Assays
Quality Control Assays
Description
Ligation Master Mix is a ready-to-use solution of T4 DNA Ligase, proprietary ligation enhancer, and
optimized reaction buffer.
Quality Control Assays
Efficiency (transformants/µg)
| Recircularization | Insertion | |
| Blunt ends | > 1 x 107 | > 2.5 x 106 |
| Uncut vector | > 1 x 108 | N/A |
Quality Control Assays
Description
The Q5 PCR Master Mix (2X) is specifically optimized for robust, high fidelity amplification of
next-generation sequencing (NGS) libraries, regardless of GC content. The polymerase component of the master
mix, Q5 High-Fidelity DNA Polymerase, is a novel thermostable DNA polymerase that possesses 3´→5´
exonuclease activity, and is fused to a processivity-enhancing Sso7d domain. Q5 also has an ultra-low error
rate (> 100-fold lower than that of Taq DNA Polymerase and ~12-fold lower than that of Pyrococcus
furiosus (Pfu) DNA Polymerase). The buffer component of the master mix has been optimized for robust
amplification, even with GC-rich amplicons and offers enhanced compatibility with a variety of beads used in
typical NGS workflows. These features make the Q5 PCR Master Mix ideal for NGS library construction. This
convenient 2X master mix contains dNTPs, Mg++ and a proprietary buffer, and requires only the addition of
primers and DNA template for robust amplification. The inclusion of the hot start aptamer allows convenient
room temperature reaction set up.
Quality Control Assays
posted November 2017
revised January 2023
protocol id: 1624
Protocol Id: 1624
Next generation sequencing (NG-seq) is a high throughput method that can be used downstream of chromatin immunoprecipitation (ChIP) and Cleavage Under Targets and Release Using Nuclease (CUT&RUN) assays to identify and quantify target DNA enrichment across the entire genome. The DNA Library Prep Kit for Illumina Systems (ChIP-seq, CUT&RUN) contains all of the enzymes and buffers necessary to generate high quality DNA sequencing libraries from ChIP DNA or CUT&RUN DNA for next-generation sequencing on the Illumina Systems platform. The fast, user-friendly workflow minimizes hands-on time needed for generation and purification of DNA libraries.
Each kit component must pass rigorous quality control standards, and for each new lot the entire set of reagents is functionally validated together by construction and sequencing of indexed libraries on the Illumina Systems sequencing platform.
This product must be used in combination with Multiplex Oligos for Illumina Systems (Single Index Primers) (ChIP-seq, CUT&RUN) #29580 or Multiplex Oligos for Illumina Systems (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538. This product provides sufficient amounts of reagents for 24 reactions and is compatible with both enzymatic- or sonication-fragmented, ChIP-enriched DNA, and CUT&RUN DNA (with different protocols for DNA enriched from ChIP or CUT&RUN assays).
Compatible Assay kits:
SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) #9003
SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) #9005
SimpleChIP® Plus Sonication Chromatin IP Kit #56383
Multiplex Oligos for Illumina Systems (Single Index Primers) (ChIP-seq, CUT&RUN) #29580
Multiplex Oligos for Illumina Systems (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538
CUT&RUN Assay Kit #86652
Non-Compatible SimpleChIP® kits:
SimpleChIP® Enzymatic Chromatin IP Kit (Agarose Beads) #9002
SimpleChIP® Plus Enzymatic Chromatin IP Kit (Agarose Beads) #9004
Note: Agarose beads are blocked with sonicated salmon sperm DNA, which will contaminate DNA
library preps and NG-seq.
Required Reagents
Reagents Included:
Reagents Not Included:
| SAFE STOP | This is a safe stopping point in the protocol, if stopping is necessary. |
When performing ChIP-seq, it is necessary to generate a control DNA sequencing library that can be used to
determine any experimental bias in DNA enrichment that is introduced during the ChIP assay and DNA library
preparation. DNA purified from input chromatin (i.e. the chromatin that was used for the IP) is typically
used to generate the control DNA library.
Starting Material: 500 pg –1 μg of ChIP DNA. To ensure optimal diversity of the DNA
sequencing libraries, we recommend using 5 ng of ChIP-enriched DNA for transcription factor or co-factor
ChIP-seq, 50 ng of ChIP-enriched DNA for total histone or histone modification ChIP-seq, and 50 ng of input
DNA for the control DNA sequencing library. If necessary, less than 5 ng of ChIP-enriched DNA can be used
for library generation; however, this may result in a lower diversity of library due to PCR bias during
amplification.
Before starting:
Before starting:
Size selection is NOT recommended during the Cleanup Adaptor-ligated ChIP DNA phase, because it results in a dramatic decrease in both yield and diversity of ChIP-seq DNA libraries.
Before starting:
Before starting:
| Reagents | Volume for 1 PCR Reaction (50 μl) |
| Purified adaptor ligated ChIP-DNA fragments (from step 9, Section III) | 15 μl |
| Q5 PCR Master Mix (•) | 25 μl |
| Single Index Primer for Illumina Systems (•) (or Dual Index 7 Primer for Illumina Systems [orange cap] for dual indexing) | 5 μl |
| Universal PCR Primer for Illumina Systems (•) (or Dual Index 5 Primer for Illumina Systems [white cap] for dual indexing) | 5 μl |
| a. | Initial Denaturation | 98°C 30 sec |
| b. | Denaturation | 98°C 10 sec |
| c. | Anneal and Extension | 65°C 15 sec |
| d. |
For starting material of 50 ng ChIP DNA, repeat steps b and c for a total of 6
cycles.
|
|
| e. | Final Extension | 65°C 3 min |
| f. | Hold | 4°C |
Before starting:
| SAFE STOP | This is a safe stopping point in the protocol, if stopping is necessary. |
When performing CUT&RUN-seq, it is necessary to generate a control DNA sequencing library that can be
used to determine any experimental bias in DNA enrichment that is introduced during the CUT&RUN assay
and DNA library preparation. DNA purified from input DNA (refer to Section V of CUT&RUN Assay Kit #86652
Protocol) is typically used to generate the control DNA library.
Starting Material: 0.5 ng–1 µg of CUT&RUN DNA. The typical yield for CUT&RUN DNA is
0.6 to 6 ng per reaction with 100,000 starting cells. To ensure optimal diversity of the DNA sequencing
libraries, we recommend using as much CUT&RUN DNA as you can obtain from a CUT&RUN reaction. If
necessary, as little as 0.1 ng of CUT&RUN DNA can be used for library generation; however, this may
result in a lower diversity of library due to PCR bias during amplification. For the control DNA sequencing
library using input DNA, 5 - 10 ng is a good starting point. Please note that a Picogreen assay is needed to
determine the concentration of CUT&RUN DNA because Nanodrop alone is not sensitive enough.
Before starting:
Before starting:
| Starting DNA | Adaptor Dilution | Working Adaptor Concentration |
| 100 ng – 1 μg | No Dilution | 15 μM |
| 5 ng – 100 ng | 1:10 | 1.5 μM |
| 2.5 ng – 5 ng | 1:25 | 0.6 μM |
| 1.25 ng – 2.5 ng | 1:50 | 0.3 μM |
| Less than 1.25 ng | 1:125 | 0.12 μM |
Size selection is NOT recommended during the Cleanup Adaptor-ligated CUT&RUN DNA phase, because it results in a dramatic decrease in both yield and diversity of DNA libraries.
Before starting:
Before starting:
| Reagents | Volume for 1 PCR Reaction (50 μl) |
| Purified adaptor ligated CUT&RUN-DNA fragments (from step 9, Section III) | 15 μl |
| Q5 PCR Master Mix ( •) | 25 μl |
| Single Index Primer for Illumina Systems ( •) (or Dual Index 7 Primer for Illumina Systems [orange cap] for dual indexing) | 5 μl |
| Universal PCR Primer for Illumina Systems (•) (or Dual Index 5 Primer for Illumina Systems [white cap] for dual indexing) | 5 μl |
| a. | Initial Denaturation | 98°C for 30 sec |
| b. | Denaturation | 98°C for 10 sec |
| c. | Anneal and Extension | 65°C for 13 sec |
| d. | For 50 - 100 ng starting CUT&RUN DNA | Repeat steps b and c for a total of 6 - 7 cycles |
| For 5 - 50 ng starting CUT&RUN DNA | Repeat steps b and c for a total of 8-12 cycles | |
| For 1 - 5 ng starting CUT&RUN DNA | Repeat steps b and c for a total of 13 - 15 cycles | |
| For 0.5 - 1 ng starting CUT&RUN DNA | Repeat steps b and c for a total of 16 - 17 cycles | |
| For 0.2 – 0.5 ng starting CUT&RUN DNA | Repeat steps b and c for a total of 18 - 19 cycles | |
| For 0.1 -0.2 ng starting CUT&RUN DNA | Repeat steps b and c for a total of 20 cycles | |
| e. | Final Extension | 65°C for 3 min |
| f. | Hold | 4°C |
Before starting:
Description
End Prep Enzyme Mix is optimized to convert 500 pg-1 μg of fragmented DNA to repaired DNA having
5´-phosphorylated, 3´-dA-tailed ends.
Quality Control Assays
Quality Control Assays
Description
Ligation Master Mix is a ready-to-use solution of T4 DNA Ligase, proprietary ligation enhancer, and
optimized reaction buffer.
Quality Control Assays
Efficiency (transformants/µg)
| Recircularization | Insertion | |
| Blunt ends | > 1 x 107 | > 2.5 x 106 |
| Uncut vector | > 1 x 108 | N/A |
Quality Control Assays
Description
The Q5 PCR Master Mix (2X) is specifically optimized for robust, high fidelity amplification of
next-generation sequencing (NGS) libraries, regardless of GC content. The polymerase component of the master
mix, Q5 High-Fidelity DNA Polymerase, is a novel thermostable DNA polymerase that possesses 3´→5´
exonuclease activity, and is fused to a processivity-enhancing Sso7d domain. Q5 also has an ultra-low error
rate (> 100-fold lower than that of Taq DNA Polymerase and ~12-fold lower than that of Pyrococcus
furiosus (Pfu) DNA Polymerase). The buffer component of the master mix has been optimized for robust
amplification, even with GC-rich amplicons and offers enhanced compatibility with a variety of beads used in
typical NGS workflows. These features make the Q5 PCR Master Mix ideal for NGS library construction. This
convenient 2X master mix contains dNTPs, Mg++ and a proprietary buffer, and requires only the addition of
primers and DNA template for robust amplification. The inclusion of the hot start aptamer allows convenient
room temperature reaction set up.
Quality Control Assays
posted November 2017
revised January 2023
protocol id: 1624
Protocol Id: 2804
Next generation sequencing (NG-seq) is a high throughput method that can be used downstream of chromatin immunoprecipitation (ChIP) and Cleavage Under Targets and Release Using Nuclease (CUT&RUN) assays to identify and quantify target DNA enrichment across the entire genome. The DNA Library Prep Kit for Illumina Systems (ChIP-seq, CUT&RUN) contains all of the enzymes and buffers necessary to generate high quality DNA sequencing libraries from ChIP or CUT&RUN DNA for next-generation sequencing on the Illumina Systems platform. The fast, user-friendly workflow minimizes hands-on time needed for generation and purification of DNA libraries. This product must be used in combination with Multiplex Oligos for Illumina Systems (Single Index Primers) (ChIP-seq, CUT&RUN) #29580 or Multiplex Oligos for Illumina Systems (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538.
This product provides sufficient amounts of reagents for 24 reactions and is compatible with both enzymatic- or sonication-fragmented, ChIP-enriched DNA. Distinct protocols are provied for DNA library preparation from ChIP and CUT&RUN DNA. This product is compatible with SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) #9003, SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) #9005, SimpleChIP® Plus Sonication Chromatin IP Kit #56383, and CUT&RUN Assay Kit #86652. This product is not compatible with SimpleChIP® Enzymatic Chromatin IP Kit (Agarose Beads) #9002 and SimpleChIP® Plus Enzymatic Chromatin IP Kit (Agarose Beads) #9004 because agarose beads are blocked with sonicated salmon sperm DNA, which will contaminate DNA library preps and NG-seq.
All Species Expected
The chromatin immunoprecipitation (ChIP) assay is a powerful and versatile technique used for probing protein-DNA interactions within the natural chromatin context of the cell (1,2). This assay can be used to identify multiple proteins associated with a specific region of the genome, or the opposite, to identify the many regions of the genome bound by a particular protein (3-6). It can be used to determine the specific order of recruitment of various proteins to a gene promoter or to "measure" the relative amount of a particular histone modification across an entire gene locus (3,4). In addition to histone proteins, the ChIP assay can be used to analyze binding of transcription factors and co-factors, DNA replication factors and DNA repair proteins. When performing the ChIP assay, cells or tissues are first fixed with formaldehyde, a reversible protein-DNA cross-linking agent that "preserves" the protein-DNA interactions occurring in the cell (1,2). Cells are lysed and chromatin is harvested and fragmented using either sonication or enzymatic digestion. The chromatin is then immunoprecipitated with antibodies specific to a particular protein or histone modification. Any DNA sequences that are associated with the protein or histone modification of interest will co-precipitate as part of the cross-linked chromatin complex and the relative amount of that DNA sequence will be enriched by the immunoselection process. After immunoprecipitation, the protein-DNA cross-links are reversed and the DNA is purified. Standard PCR or Quantitative Real-Time PCR can be used to measure the amount of enrichment of a particular DNA sequence by a protein-specific immunoprecipitation (1,2). Alternatively, the ChIP assay can be combined with genomic tiling micro-array (ChIP on chip) techniques, high throughput sequencing, or cloning strategies, all of which allow for genome-wide analysis of protein-DNA interactions and histone modifications (5-8).
Except as otherwise expressly agreed in a writing signed by a legally authorized representative of CST, the following terms apply to Products provided by CST, its affiliates or its distributors. Any Customer's terms and conditions that are in addition to, or different from, those contained herein, unless separately accepted in writing by a legally authorized representative of CST, are rejected and are of no force or effect.
Products are labeled with For Research Use Only or a similar labeling statement and have not been approved, cleared, or licensed by the FDA or other regulatory foreign or domestic entity, for any purpose. Customer shall not use any Product for any diagnostic or therapeutic purpose, or otherwise in any manner that conflicts with its labeling statement. Products sold or licensed by CST are provided for Customer as the end-user and solely for research and development uses. Any use of Product for diagnostic, prophylactic or therapeutic purposes, or any purchase of Product for resale (alone or as a component) or other commercial purpose, requires a separate license from CST. Customer shall (a) not sell, license, loan, donate or otherwise transfer or make available any Product to any third party, whether alone or in combination with other materials, or use the Products to manufacture any commercial products, (b) not copy, modify, reverse engineer, decompile, disassemble or otherwise attempt to discover the underlying structure or technology of the Products, or use the Products for the purpose of developing any products or services that would compete with CST products or services, (c) not alter or remove from the Products any trademarks, trade names, logos, patent or copyright notices or markings, (d) use the Products solely in accordance with CST Product Terms of Sale and any applicable documentation, and (e) comply with any license, terms of service or similar agreement with respect to any third party products or services used by Customer in connection with the Products.