| Cat. # | Size | Qty. | Price |
|---|---|---|---|
| 51660S | 100 µl |
|
| REACTIVITY | All |
| SENSITIVITY | Endogenous |
| MW (kDa) | |
| Source/Isotype | Mouse IgG1 |
Product Information
| Application | Dilution |
|---|---|
| Immunofluorescence (Immunocytochemistry) | 1:400 |
| DNA Dot Blot | 1:1000 |
IMPORTANT: This protocol employs an atypical fixation and denaturation strategy with which only certain targets are compatible. Where appropriate, this protocol will be linked to its validated antibody under the Product Information banner on the product-specific webpage.
NOTE: Prepare solutions with reverse osmosis deionized (RODI) or equivalently purified water.
NOTE: All subsequent incubations should be carried out at room temperature (20-25°C) unless noted otherwise.
Rinse three times in 1X PBS for 5 min each.
NOTE: If using a fluorochrome-conjugated primary antibody, then skip to Section C, Step 8.
Counterstain as appropriate.
NOTE: When including fluorescent cellular dyes in your experiment (DNA dyes, etc.), please refer to the dye product page for its recommended protocol. View our listing of cellular dyes validated for use in immunofluorescence.
posted December 2015
revised December 2020
Protocol Id: 865
Note: This protocol is written for spotting fragmented, purified genomic DNA (titration of 1000 ng, 500 ng, 250 ng, 125 ng, 62.5 ng, 31.25 ng, and 15.625 ng) onto a positively charged nylon membrane using a 96-well dot blotting apparatus. Depending on the source and type of DNA, more or less DNA may be required for detection with the antibody.
Before Starting:
• Purify genomic DNA using a genomic DNA purification protocol or kit and sonicate
genomic DNA to generate fragments between 200 and 500 bp. DNA fragment size
can be analyzed by gel electrophoresis on a 1% agarose gel with a 100 bp DNA
marker.
• Cut a piece of nylon membrane to the size of the dot blot manifold.
• Wet nylon membrane with 10x SSC Buffer.
• Dry membrane by placing it in 96-well dot blot apparatus and applying vacuum.
NOTE: Due to the kinetics of the detection reaction, signal is most intense immediately following incubation and declines over the following 2 hr
posted November 2015
Protocol Id: 804
All Species Expected
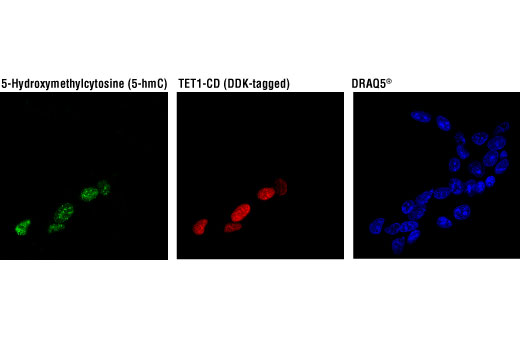
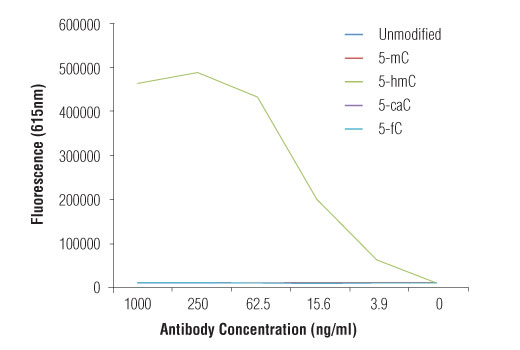
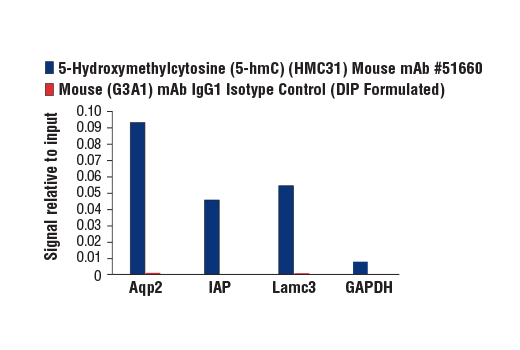
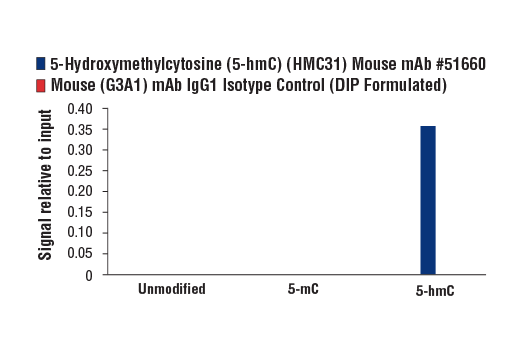
Monoclonal antibody is produced by immunizing animals with 5-hydroxymethylcytidine.
Methylation of DNA at cytosine residues is a heritable, epigenetic modification that is critical for proper regulation of gene expression, genomic imprinting, and mammalian development (1,2). 5-methylcytosine is a repressive epigenetic mark established de novo by two enzymes, DNMT3a and DNMT3b, and is maintained by DNMT1 (3, 4). 5-methylcytosine was originally thought to be passively depleted during DNA replication. However, subsequent studies have shown that Ten-Eleven Translocation (TET) proteins TET1, TET2, and TET3 can catalyze the oxidation of methylated cytosine to 5-hydroxymethylcytosine (5-hmC) (5). Additionally, TET proteins can further oxidize 5-hmC to form 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC), both of which are excised by thymine-DNA glycosylase (TDG), effectively linking cytosine oxidation to the base excision repair pathway and supporting active cytosine demethylation (6,7).
TET protein-mediated cytosine hydroxymethylation was initially demonstrated in mouse brain and embryonic stem cells (5, 8). Since then this modification has been discovered in many tissues, with the highest levels found in the brain (9). While 5-fC and 5-caC appear to be short-lived intermediate species, there is mounting evidence showing that 5-hmC is a distinct epigenetic mark with various unique functions (10,11). The modified base itself is stable in vivo and interacts with various readers including MeCP2 (11,12). The global level of 5-hmC increases during brain development and 5-hmC is enriched at promoter regions and poised enhancers. Furthermore, there is an inverse correlation between levels of 5-hmC and histone H3K9 and H3K27 trimethylation, suggesting a role for 5-hmC in gene activation (12). Lower amounts of 5-hmC have been reported in various cancers including myeloid leukemia and melanoma (13,14).
Except as otherwise expressly agreed in a writing signed by a legally authorized representative of CST, the following terms apply to Products provided by CST, its affiliates or its distributors. Any Customer's terms and conditions that are in addition to, or different from, those contained herein, unless separately accepted in writing by a legally authorized representative of CST, are rejected and are of no force or effect.
Products are labeled with For Research Use Only or a similar labeling statement and have not been approved, cleared, or licensed by the FDA or other regulatory foreign or domestic entity, for any purpose. Customer shall not use any Product for any diagnostic or therapeutic purpose, or otherwise in any manner that conflicts with its labeling statement. Products sold or licensed by CST are provided for Customer as the end-user and solely for research and development uses. Any use of Product for diagnostic, prophylactic or therapeutic purposes, or any purchase of Product for resale (alone or as a component) or other commercial purpose, requires a separate license from CST. Customer shall (a) not sell, license, loan, donate or otherwise transfer or make available any Product to any third party, whether alone or in combination with other materials, or use the Products to manufacture any commercial products, (b) not copy, modify, reverse engineer, decompile, disassemble or otherwise attempt to discover the underlying structure or technology of the Products, or use the Products for the purpose of developing any products or services that would compete with CST products or services, (c) not alter or remove from the Products any trademarks, trade names, logos, patent or copyright notices or markings, (d) use the Products solely in accordance with CST Product Terms of Sale and any applicable documentation, and (e) comply with any license, terms of service or similar agreement with respect to any third party products or services used by Customer in connection with the Products.