The SignalScan Peptide Mix (SARS-CoV-2) enables highly sensitive, consistent profiling of disease-relevant peptides for quantification of protein levels as well as post-translational modifications in unenriched samples. Used with SISAS or PRM/MRM LC-MS analytical methods, the peptide mix delivers multiplexed, accurate analysis of viral and host immune response proteins.
To quickly work toward a better understanding of SARS-CoV-2 viral entry and host immune response, researchers are using a wide variety of analytical techniques. Monitoring numerous viral and immune protein targets via individual assays, like western blot or ELISA, is a lot of work and requires a large sample amount. By using specific targeted proteomics techniques, you can zero in on preselected targets and quantify post-translational changes in many proteins at once. This targeted proteomics approach, with inherent multiplexing capability, not only saves valuable time and effort, it results in more focused, quantitative data to support complex hypotheses. The SISAS workflow, a targeted LC-MS/MS-based assay, offers efficient, sensitive, and highly specific analysis of many relevant SARS-CoV-2 proteins and post-translational modifications (PTMs) in a single injection.
The SignalScan Peptide Mix (SARS-CoV-2) is a synthesized mixture of isotopically labeled (13C & 15N) peptides and phosphopeptides that have been carefully selected to represent 11 highly relevant proteins-of-interest for SARS-CoV-2 infection and human innate immune response. The mixture includes 96 pmol of each peptide and phosphopeptide, which are stable-isotope labeled on the carboxy-terminal amino acid (15N413C6-Arg or 15N213C6-Lys) and quantified by amino acid analysis (AAA). Many of these stable isotope standard (SIS) peptides include important post-translational modifications derived from Nucleocapsid and innate immune response proteins, as well as Spike cleavage variants, that can be measured to reveal changes in the activity of SARS-CoV-2 proteins and immune signaling pathways.
| Protein | Phosphorylation site | Function |
|---|---|---|
| Nucleocapsid | S176, T198, S206 | Viral assembly |
| Spike | Cleaved & uncleaved R815 | Viral assembly |
| ACE2 | Viral entry | |
| IL6 | Innate immune response | |
| IL8 | Innate immune response | |
| Interferon alpha | Interferon signaling | |
| Interferon beta | Interferon signaling | |
| STAT1 | S727, Y701 | Interferon signaling |
| Jak1 | Y1034, Y1035 | Interferon signaling |
| PERK | S715 | Translational activity |
| IRAK4 | T345, S346 | TLR activation/viral RNA detection |
The SISAS workflow enables you to perform a deep analysis of the target peptides from human samples and compare results across infection and treatment conditions. After endogenous cellular protein has been extracted, it is digested into peptides with trypsin. The SignalScan™ Peptide Mix (SARS-CoV-2) can then be spiked into a sample of test peptides before performing LC-MS/MS analysis, using SISAS or PRM/MRM methods.

The SISAS (Stable-Isotope-Standard-Assisted Scanning) workflow is a tandem mass spectrometry technique in which spiked-in standards of preselected heavy isotope-labeled peptides are detected via a fast MS2 scan to simultaneously focus on matching endogenous peptides. Once the target peptides are identified using a MS2 survey scan in “Watch Mode,” the LC-MS2 program switches to “Quantification Mode“ to perform high-resolution MS2 scans that enable high-quality quantification of the target elution peak. This method can be performed using HRAM Orbitrap LC-MS systems, including Thermo Scientific Orbitrap Exploris 480, Orbitrap Eclipse Tribrid, and other Tribrid Orbitrap MS systems with Tune v3.3 or higher.

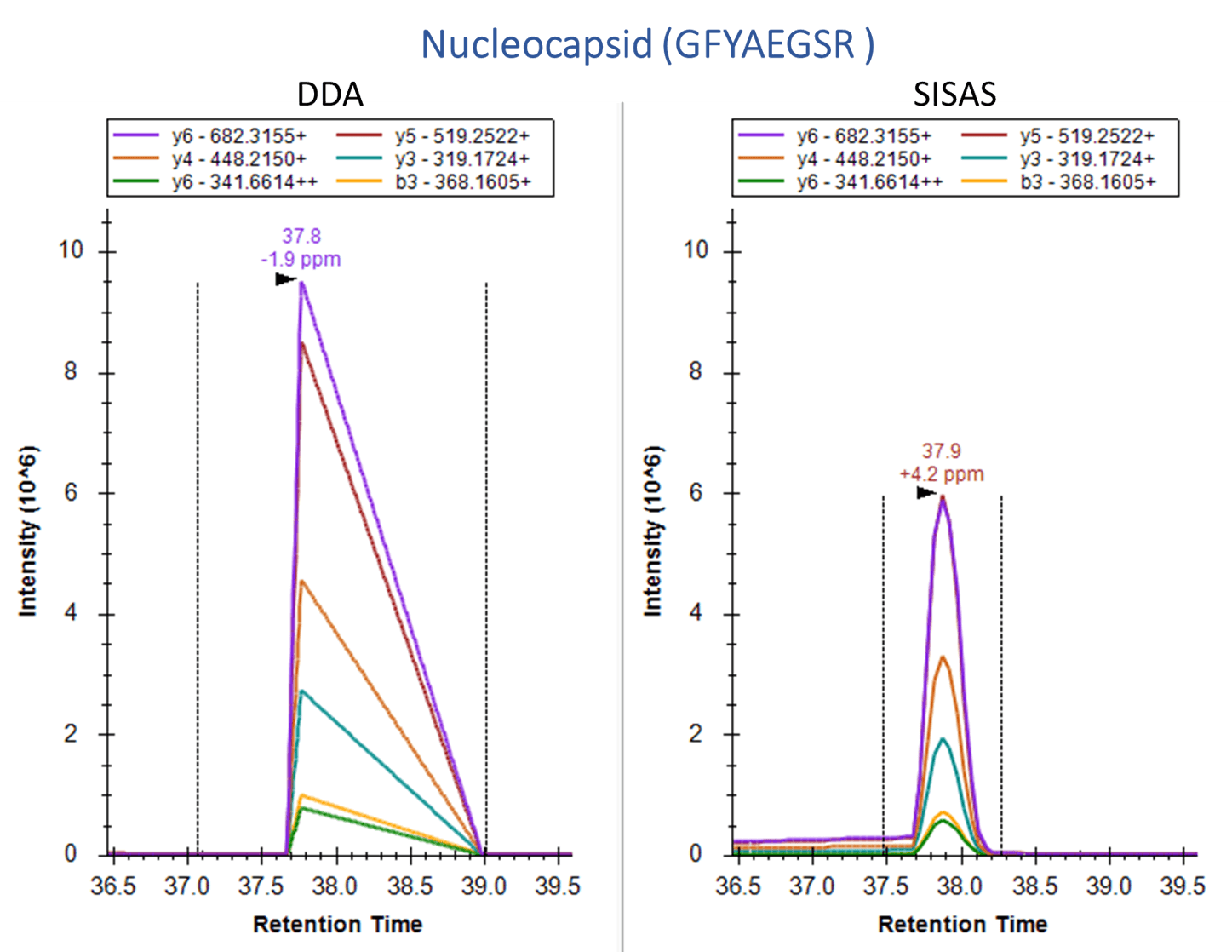
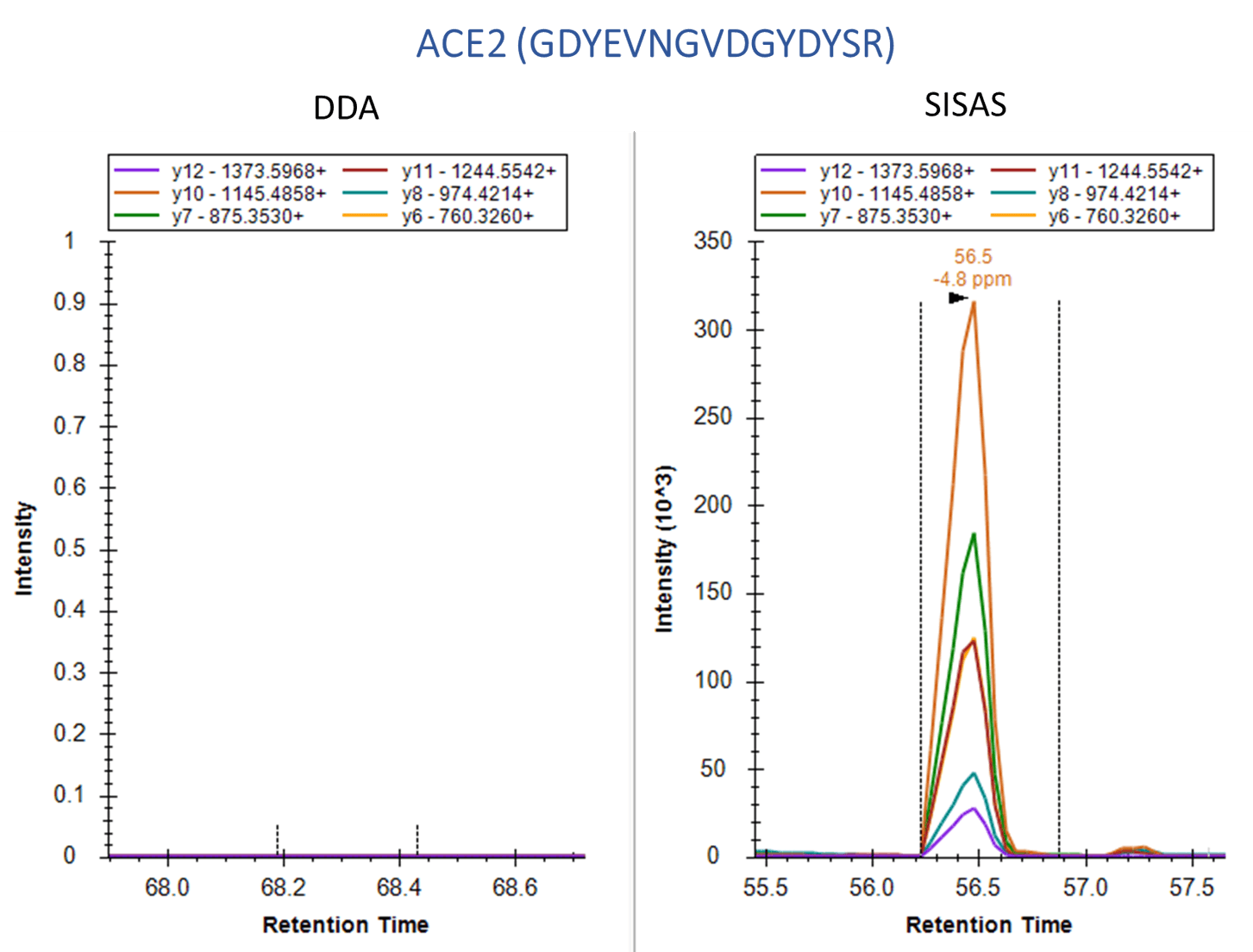
Human A549 cells, which overexpress the ACE2 receptor, were examined using the SignalScan Peptide Mix (SARS-CoV-2) and SISAS workflow. The following are product ion chromatograms, visualized in Skyline software, of peptides from the SARS-CoV-2 Nucleocapsid protein (top) and the human ACE2 protein (bottom), derived from virally infected A549 cells. Using a standard data-dependent analysis (DDA) LC-MS/MS method (left panels) results in no signal or collapsed peaks. The Stable-Isotope-Standard-Assisted Scanning (SISAS) method (right panels) results in consistently detected peptides with clear peaks, resulting in confident peptide and protein quantification.
The SignalScan Peptide Mix (SARS-CoV-2) and SISAS workflow was used to examine human A549 cells that overexpress the ACE2 receptor. The cells were either mock-infected or infected with SARS-CoV-2, and infected cells were either untreated (labeled SARS-CoV-2) or treated with a kinase inhibitor. Here we present a heatmap of peptide abundance of all SignalScan Peptide Mix (SARS-CoV-2) targets in these cells. Biological triplicates of different infection and treatment conditions cluster together, as do many related peptides, including those derived from viral proteins. For heatmap generation, raw intensities were normalized to the maximum observation among all samples for each peptide. Each sample was clustered using the unweighted pair group method with arithmetic mean (UPGMA) algorithm, and a dendrogram was generated from this clustering and plotted with a heatmap of normalized intensities.
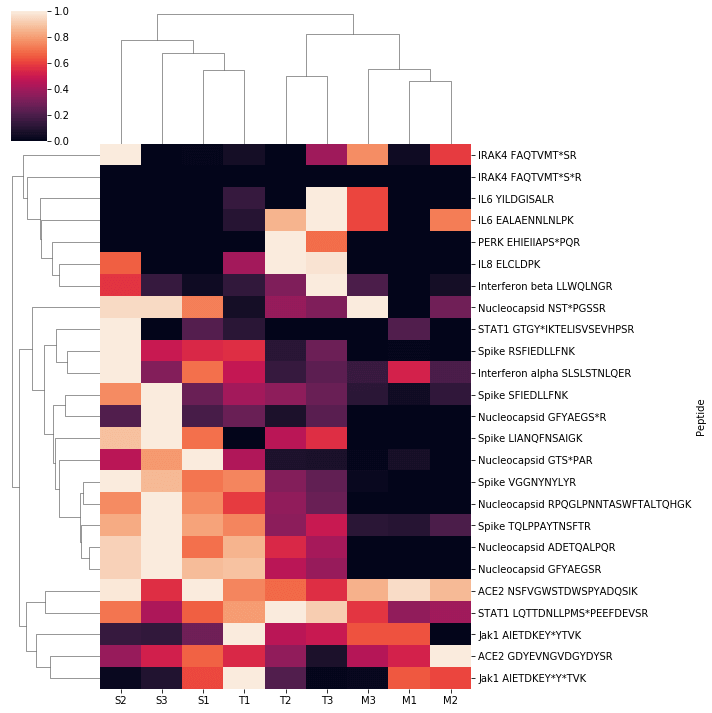
See the product SignalScan Peptide Mix (SARS-CoV-2).