

Overview of Chromatin Immunoprecipitation (ChIP)
1. What is ChIP?
Chromatin immunoprecipitation, or ChIP, is an antibody-based technology used to selectively enrich specific DNA-binding proteins along with their DNA targets. ChIP is used to investigate a particular protein-DNA interaction, several protein-DNA interactions, or interactions across the whole genome or a subset of genes.
ChIP utilizes antibodies that selectively recognize and bind proteins, including histones, histone modifications, transcription factors, and cofactors, to provide information about chromatin states and gene transcription. A combination of proteomic analysis and molecular biology techniques used in ChIP allow for the ability to understand gene expression and regulation in cells or tissues of interest.
2. When is ChIP used?
Typically, ChIP is used to identify the relative abundance of a specific protein or a specific protein modification at a certain region in the genome. ChIP can be used to answer a multitude of scientific questions involving the interaction of proteins and chromatin. For example, ChIP can be used to compare the presence of certain proteins at various loci, map the various proteins across a genomic region of interest, or quantify protein binding to an inducible gene in response to a stimulus over time.
3. How does ChIP work?
The principle behind ChIP is relatively straightforward and relies on the use of an antibody to isolate, or precipitate, a certain protein, histone, transcription factor, or cofactor and its bound chromatin from a protein mixture that was extracted from cells or tissues. Hence, the name of the technique: Chromatin Immunoprecipitation. In ChIP-PCR or ChIP-seq, immune-enriched DNA fragments are then able to be identified and quantified using widely available PCR or qPCR reagents and Next Generation Sequencing (NGS) technologies.
4. What is native ChIP (N-ChIP) vs crosslinked ChIP (X-ChIP)?
There are 2 types of ChIP techniques that can be carried out depending on the experimental question and the starting material for the experiment: 1) native ChIP (N-ChIP) and 2) crosslinked ChIP (X-ChIP). Both types of ChIP have advantages and disadvantages:
- In N-ChIP, no fixing agent is used to crosslink proteins to the chromatin. Instead, native chromatin is isolated from cell nuclei that are digested with a nuclease. Because antibodies are raised against unfixed antigens, N-ChIP offers the advantage of better recognition and binding of antibodies to their target antigens. PCR may not be required for downstream analysis due to the high abundance of histone proteins. While these advantages make N-ChIP an attractive method, it can only be used for the detection of histones. In addition, loss of protein binding during the chromatin digestion and immunoprecipitation steps may bias the data or impede proper analyses.
- In X-ChIP, chemical fixatives such as formaldehyde are used to crosslink the protein of interest to the DNA and fragmentation of chromatin is achieved through sonication or nuclease digestion. The advantage of X-ChIP is that it can be used with histone and non-histone proteins and generally requires less cellular starting material than N-ChIP. X-ChIP also minimizes the chances of chromatin protein loss during extraction, allowing for the detection of transient protein interactions. However, the precipitation step is less efficient and DNA amplification by PCR is necessary for downstream analyses.
5. What are the different types of ChIP assays?
Once the chromatin immunoprecipitation itself is complete, several downstream analyses can be conducted on the purified chromatin and the associated proteins, histones, transcription factors, and cofactors. The most common methods for single gene analysis and whole genome analysis are qPCR and ChIP-seq, respectively. PCR and ChIP-chip are also options for downstream analysis.
5.1 What are the advantages of ChIP-PCR?
ChIP-PCR is performed to analyze histone modifications and/or protein binding to a known subset of target loci in the genome. In ChIP-PCR, immune-enriched DNA fragments are identified and quantified using widely available PCR or qPCR reagents and technologies. Rapid and quantitative comparisons of specific regions within the genome across multiple samples can be achieved using ChIP-qPCR. This is cheaper and more time efficient than whole genome sequencing methods.
5.2 What are the advantages of ChIP-chip?
ChIP-chip technology refers to the utilization of a DNA microarray chip to analyze ChIP-immune enriched DNA fragments. Using genome tiling microarray technology allows for a whole-genome analysis of proteins that are bound to isolated DNA and generates a high resolution genomic map of protein-binding and protein modifications. ChIP-chip has multiple uses in basic research as well as disease-based research. For example, it can be used to identify the binding sites of transcription factors, enhancers, and repressors and to compare these types of bound proteins in control and pathological samples. However, since the cost of NGS has decreased substantially and similar results are obtained using ChIP-seq, more people are choosing to perform ChIP-seq instead of ChIP-chip.
5.3 What are the advantages of ChIP-seq?
Similar to ChIP-chip, ChIP-seq provides information about genome-wide protein binding. However, unlike ChIP-chip, ChIP-seq uses NGS technology to identify DNA fragments and map them against the entire genome.
More contemporary DNA amplification technology allows for robust analysis to be conducted in a matter of days with a low amount of input DNA. When starting material is scarce, these technological advancements in library preparation methods have made ChIP-seq experiments possible.
In addition, new technology in which DNA samples are uniquely tagged with short sequences, known as barcodes, now allows individual fragments to be pooled into a single sequencing lane for multiplexed analysis. This has substantially increased efficiency and decreased cost of DNA sequencing experiments, further supporting ChIP-seq applications.
Taken together, due to advancements in DNA-sequencing technology, the advantage of ChIP-seq is that a large number of ChIP-enriched DNA samples can be sequenced cheaply in a relatively short period of time with higher sensitivity and accuracy than ChIP-chip.
6. What are the different steps in the ChIP assay?
ChIP assays follow a general protocol:
- Crosslinking of proteins to DNA for X-ChIP only
- Cell lysis
- Chromatin fragmentation by digestion (for X-ChIP and N-ChIP) or sonication-based shearing (for X-ChIP only)
- Immunoprecipitation using specific antibodies
- DNA clean-up for downstream analyses
- DNA analysis via PCR, qPCR, microarray, or NGS
Importantly, positive and negative controls at each step are integral for determining whether a ChIP experiment has been successful.
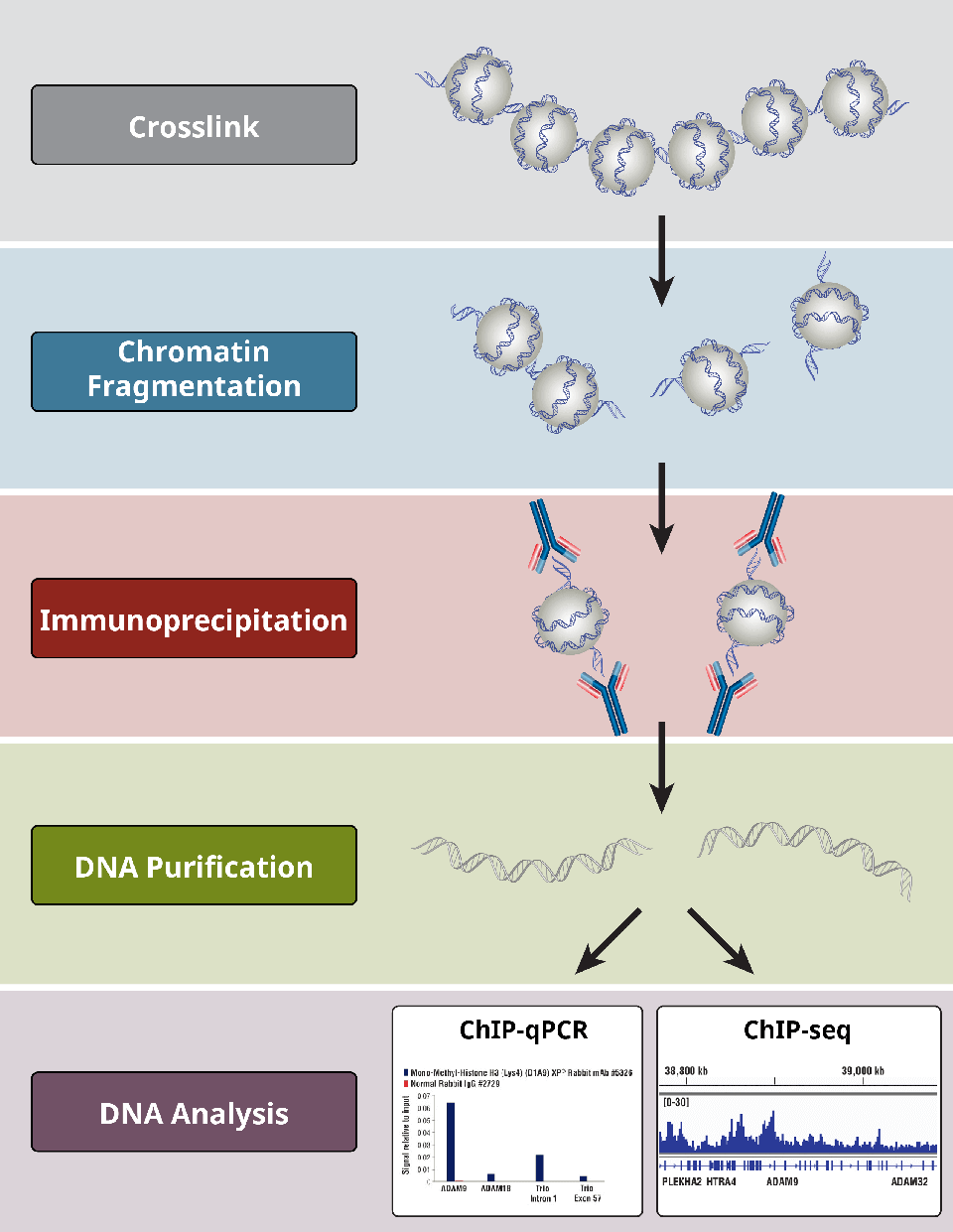
Overview of the most critical steps of a ChIP protocol.
6.1 How do you crosslink cells and tissues for ChIP?
Crosslinking reagents are used to “fix” proteins to the DNA that they bind. Formaldehyde-based reagents are typically used to achieve this fixation. Cells and tissues are generally fixed in a similar manner, but tissue requires a longer fixation time and a more rapid fixation delivery in order to quickly permeate the target tissue before it begins to degenerate.
Over-fixation of chromatin can reduce the efficiency of fragmentation by sonication, in addition to inhibiting the binding of antibodies to their protein targets. Therefore, fixation time should be empirically determined in order to allow for maximal antibody-antigen binding while achieving ideal crosslinking of proteins to their target DNA.
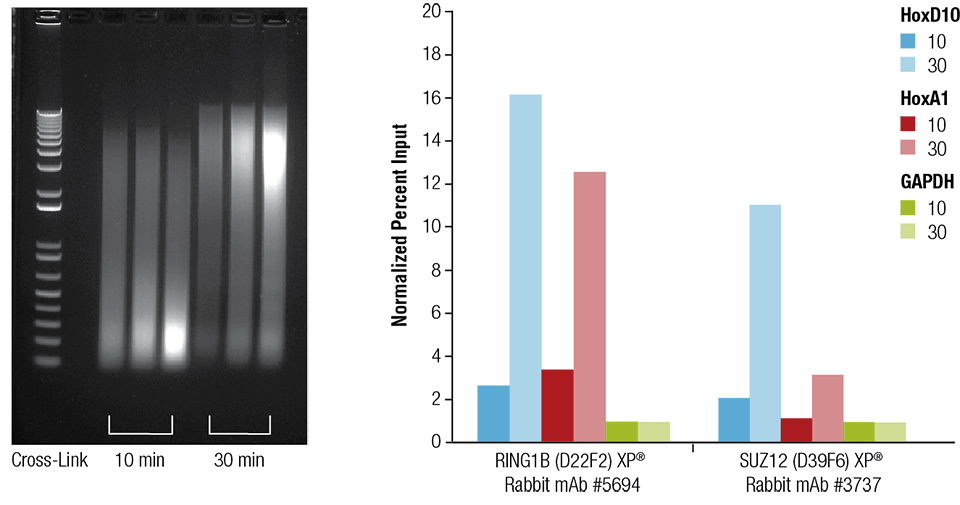
Mouse heart (H), brain (B), and liver (L) were crosslinked for 10 or 30 minutes as indicated (left panel). The chromatin was prepared and sonicated for 4 minutes. ChIP was performed using chromatin prepared from heart tissue with the indicated antibodies using the SimpleChIP® Plus Sonication Chromatin IP Kit #56383 and the enriched DNA was quantified by real-time PCR using primers to the indicated genes (right panel). The amount of immunoprecipitated DNA in each sample is represented as normalized signal to the negative loci GAPDH, which is equal to 1.
6.2 How do you fragment chromatin?
Chromatin fragmentation is essential for the success of a ChIP experiment. Fragmentation of chromatin is necessary to solubilize the chromatin and allows for its coprecipitation. In addition, the resolution of the ChIP assay depends on the fragmentation of the chromatin, since DNA fragment size determines the resolution of the ChIP assay.
Enzymatic digestion uses micrococcal nuclease (MNase), which cleaves double-stranded DNA between nucleosomes to generate chromatin fragments. While a complete MNase digestion generates DNA fragments of 150 base pairs (mono-nucleosomes), incomplete digestion generates DNA fragments between 150 and 750 base pairs (mono-, di-, and tri-nucleosomes). Sonication uses mechanical force to fragment chromatin. Sonication fragments chromatin between and within nucleosomes, generating a range of chromatin fragments between 150 and 1000 base pairs.
For X-ChIP, either enzymatic digestion or sonication is used to shear the chromatin. Sonication conditions in the sonication ChIP protocol should be empirically determined, as they differ by cell type and experimental condition. Digestion conditions are more consistent across different cell types and tissues, but chromatin fragment size should still be analyzed prior to IP.
For N-ChIP, nucleases are used to fragment the chromatin in order to maintain protein binding in unfixed samples. Nuclease fragmentation should also be empirically determined to minimize overdigestion of the chromatin.
6.3 Why use enzymatic digestion for ChIP?
Nuclease digestion must be used for N-ChIP since proteins are not crosslinked to the DNA and the harsh conditions associated with sonication-based fragmentation would result in dissociation of chromatin proteins from the DNA. N-ChIP is ideal for analyzing histone protein-DNA interactions, because histone-DNA binding is very strong and stable. However, N-ChIP does not work well for analysis of transcription factor and cofactor chromatin binding.
Either enzymatic digestion or sonication can be used to fragment chromatin in X-ChIP. The benefits of enzymatic digestion include the consistency of fragmentation and mild fragmentation conditions (lower heat and detergent) that better preserve the integrity of the chromatin and antibody epitopes, resulting in increased immune-enrichment of transcription factor and cofactor bound chromatin.
6.4 Why use sonication to fragment chromatin for ChIP?
Unlike the chromatin fragmentation achieved through enzymatic digestion, sonication relies on mechanical forces to fragment chromatin into smaller pieces. The ideal size of chromatin fragments for immune-enrichment is between 200 and 1000 base pairs. Sonication is the traditional method used to fragment chromatin and can be performed using a traditional probe sonicator or more advanced water-bath sonicators that provide more focused sonication. Sonication generates truly randomized fragments of chromatin; however, it requires extensive optimization across different cell lines and tissues and is difficult to reproduce from experiment to experiment. The requirement for high detergent buffers and the heat generated during sonication can damage the integrity both of the chromatin and antibody epitopes on chromatin proteins.
6.5 Optimizing chromatin sonication for ChIP
Sonication-based chromatin fragmentaton traditionally uses high detergent buffers and generates heat, both of which can damage the integrity of the chromatin and antibody epitopes. Therefore, the amount of sonication used to fragment chromatin must be experimentally determined for different cell lines and tissues. One must identify and use the minimal amount of sonication required to generate 150 to 1000 base pair DNA fragments to minimize damage to the chromatin.
Before embarking on a full ChIP assay with downstream analysis by qPCR, DNA chip, or NGS, gel electrophoresis should be used to analyze chromatin samples sonicated for various times. Fragment size depends on sonication time-fragment size decreases as sonication time increases. However, data suggest that longer sonication times does not lead to better results. Therefore, running purified immunoprecipitated DNA on a gel and determining the ideal fragment size is a straightforward method for determining the minimal amount of sonication required for the desired DNA size and to avoid unnecessary damage to chromatin.

Enzyme-digested chromatin was run on an agarose gel. Lane 1 shows chromatin that is underdigested. Lane 2 shows properly digested chromatin, and Lane 3 shows chromatin that is overdigested.
6.6 How do you choose an antibody for ChIP?
Choosing an appropriate antibody for a ChIP experiment is integral to its success. Antibodies used in a ChIP experiment should be specific for the protein of interest and have high affinity for the antigen. The best choice of antibody for a ChIP or ChIP-seq experiment is a ChIP- or ChIP-seq-validated antibody. If there is no ChIP-validated antibody available for the gene of interest, the next best choice is an antibody that has been validated in IP. It is important to note that not all IP-validated antibodies work in ChIP and not all ChIP-validated antibodies work in ChIP-seq. In addition, the more an antibody is validated across other applications, such as western, IP, IF, flow, and IHC, the more confidence one can have in antibody performance and specificity. The validation of an antibody must be followed by empirical determination of the optimal concentration of the antibody, along with IP washing conditions.
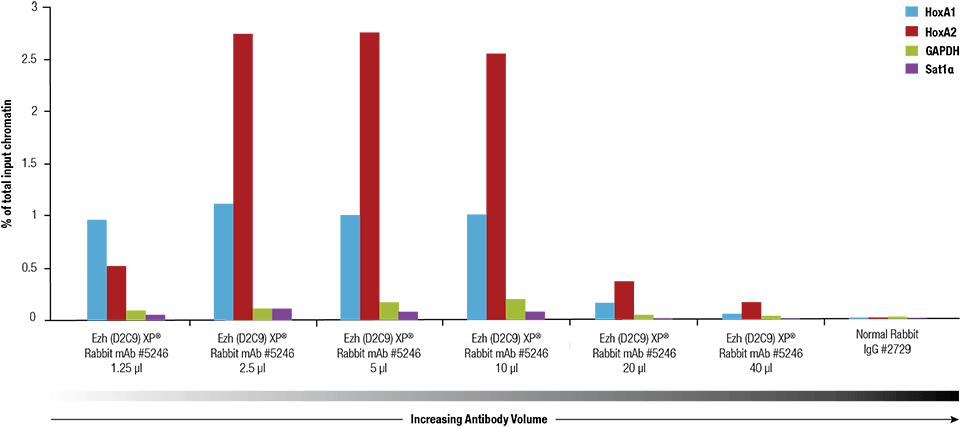
Ezh2 (D2C9) XP® Rabbit mAb #5246 was titrated using the SimpleChIP® Plus Enzymatic Chromatin IP Kit #9005 on crosslinked chromatin prepared from 4 x 106 NCCIT cells.
6.7 How to perform the immunoprecipitation
Antibodies are used to capture a protein of interest and its bound DNA. Antibody concentration should be determined empirically, with a general starting point of 0.5-2.0 μg of antibody used per 10 μg of chromatin DNA (corresponds to approximately 4 x 106 cells). The stringency of buffers and wash times should also be empirically determined as they depend on the affinity of the antibody to its target antigen. Typically, antibody:chromatin incubations are done for 2 hours to overnight.
Antibody-antigen (+DNA) complexes are affinity captured on an antibody-binding resin. In ChIP experiments this resin is typically composed of ChIP-grade magnetic, sepharose, or agarose beads conjugated to protein G. Antibodies bind protein A and/or protein G beads with various affinities depending on the species in which they were developed and the IgG subtype of their heavy chain. Beads are typically incubated with antibody:chromatin for 2 to 4 hours.
Wash steps are needed to remove non-antibody-bound chromatin, followed by reversal of crosslinks (for X-ChIP) and purification of DNA. In addition, an IgG control IP must be performed to determine background (signal:noise). Positive control antibodies (ie. total histone H3) and/or positive control qPCR primers (for known positive and negative target protein binding loci) must also be included to determine non-specific binding. For optimal results, QC of the chromatin IP by qPCR should be performed prior to downstream NGS analysis.
6.8 How to elute the chromatin from the protein A/G beads
Chromatin is eluted from the protein A/G beads using detergent and heat. Low-speed "vortexing" or mixing is required to keep the beads in suspension and to increase elution of the chromatin.
6.9 How to reverse the chromatin crosslinks
Crosslinks are reversed by high heat and high salt (both of these are vital components). Proteinase K is also added to digest the associated chromatin proteins and added antibodies, allowing for more efficient downstream DNA purification.
6.10 How to purify DNA
After the chromatin crosslinks are removed, DNA is either purified using classic phenol-chloroform followed by ethanol precipitation methods or using column-based DNA purification kits.
7. How to analyze enriched DNA
Once DNA is purified, several downstream analyses can be conducted, including ChIP-PCR, ChIP-qPCR, ChIP-chip, and ChIP-seq.
7.1 ChIP-PCR and ChIP-qPCR analysis
ChIP-PCR and ChIP-qPCR analyses are best for single-gene analysis and can be used to amplify and quantify specific fragments of DNA in a rapid and cost-effective manner.
7.2 ChIP-chip analysis
ChIP-chip analysis utilizing tiling DNA microarray chips to create a genome-wide, high resolution map of protein binding and protein modification.
7.3 ChIP-seq analysis
ChIP-seq analysis uses standard NGS technology to align purified DNA with previously annotated whole genomes to identify genome-wide protein binding profiles.