

Overview of Enzyme-Linked Immunosorbent Assay (ELISA)
What Is an Enzyme-Linked Immunosorbent Assay (ELISA)?
An enzyme-linked immunosorbent assay (ELISA) is a qualitative or quantitative test that uses antibodies to bind and measure a molecule of interest. Similar to other immunoassays, both monoclonal and polyclonal antibodies can be utilized to identify the analyte (eg, peptides, proteins, antibodies, small molecules). The antibody provides the specificity for the analyte, and a moiety, such as horseradish peroxidase (HRP) is either directly or indirectly coupled to the antibody in order to provide the detection method and possible signal amplification. Multiple ELISA methods and detection chemistries allow the end user to fit the proper assay to the intended experimental question. The format can be used as both a 1-sample test or a high-throughput method for screening.
ELISA Method and Principle
The experimental setup will vary depending on the exact type of ELISA being used (see below for detailed descriptions of various types of ELISAs). One of the simplest forms of an ELISA begins with immobilizing the antigen of interest on a solid surface. This is typically done in a 96- or 384-well plate that passively binds proteins. The appropriate antibody is prepared by conjugation to an enzyme. This conjugate is added to the sample allowing the antibody to bind the antigen. Bound material that is nonspecific to the antigen of interest can then be removed using washing steps. Substrate for the enzyme is then added so that an enzymatic reaction between the substrate in the solution and the antibody-linked enzyme takes place, ultimately producing either a change in color, fluorescence, or luminescence that is used as a readout to determine the quantity of the target analyte in each sample.
What Does ELISA Detect and Measure?
An ELISA is an indirect measure of the binding of antibody with antigen. The quality of the assay will depend in large part on the specificity of the antibody for the antigen. If the specificity is poor, there will be a highly nonspecific background. If the binding is specific but weak, the wash steps will remove some of the antibody, leading to a falsely low signal. The detection of the binding is indirect, since the signal is generated from an enzyme conjugated to an antibody. This is a relative signal and may depend on incubation times and temperatures. Quantitation can be achieved through standard curves if available.
Applications of ELISA
ELISAs are used to quantify 1 or more antigens in a test sample. Because of the specificity of the antibody reagents and the simplicity of the test, an ELISA is typically considered as the gold standard for quantitative testing of biologic samples. Tests that use the ELISA principle of antibody-antigen binding with amplification of signal are ubiquitous in the biomedical/biochemical fields. In the research lab, these tests can be used to quantitate protein levels or pathway activation. In the clinic, ELISAs have a variety of uses, including measuring biomarkers from drug treatments or levels of cytokines in patient serum samples. ELISAs are also applicable in plant biology and can be used in tests for crop allergens.
Using ELISA as a Research Tool
The ELISA platform is used in multiple research areas. Because of its ability to detect antibodies specifically and sensitively, it is a convenient tool for biological discovery. It is broadly used across all biologic research areas to detect and quantitate antigens of interest. The antigens could be secreted into cell culture media, total proteins in a cell, or post translational modifications (PTMs) that occur during cell signaling.
Using ELISA as a Diagnostic Tool
Along with its use in research, ELISAs are broadly used for diagnostic testing, including for viral infections, food allergens, toxicology, and cancer. Detection of the infectious virus by antibody provides a rapid clinical test that is scalable to testing many samples in parallel. These assays are typically completed on serum or blood samples, but could also be performed using lysates of cells or tissues.
Benefits of ELISA
ELISA tests typically use basic mix and wash protocols and simple equipment, making them easy to perform in the lab. The key reagent, the antibody, binds specifically to its target with a low off rate, offering results with high sensitivity and specificity. Additionally, data are generally straightforward to analyze, they can be quantitative or qualitative depending on experimental set-up, and results are clear and easy to interpret.
ELISA detection for lab tests requires equipment typically found in most research and clinical laboratories (plate-reader capable of measuring absorbance, fluorescence, and/or chemiluminescence). For other applications, the readout can be colored spots or lines. ELISA assays are also scalable and can be built in a variety of formats, from 96 or 384 well plates in the lab to over-the-counter pregnancy tests. Another advantage of ELISA tests is that validated antibodies for ELISA can be reproduced in large scale, providing a robust source of reagent for long-term use.
Types of ELISA Methods
There are several types of commonly used methods for ELISA. From these methods, many other variations have been used depending on the biological question, available binder, and detection methodology:
- Direct ELISA – Enzyme conjugated to antibody that binds to antigen on a surface.
- Indirect ELISA – Similar to direct ELISA, but the antibody is not conjugated. A second conjugated antibody is used to detect the bound antibody.
- Sandwich ELISA – The antigen is recognized by 2 antibodies, making a complex like a sandwich. One antibody is used for capture and one is used for detection. The detection can be direct or indirect.
- Competition/inhibition ELISA – Similar to a direct ELISA, but the quantitation is performed by competing or inhibiting the antibody binding with a measured amount of antigen.
It should be noted that the terms “direct/indirect” and “primary/secondary” have different meanings in the literature, depending on context. The “direct” ELISA has been used to mean directly binding antigen to a surface then detecting with antibody. However, “direct” has also been used to describe binding an antibody directly to any antigen regardless of format. The “indirect” ELISA terminology refers to an extension of the direct method but there is binding of a second antibody to the first antibody. “Indirect” can also refer more generally to any scheme where detection comes from a second antibody, not the antibody that binds the antigen of interest. The terminology of “primary” refers to the binding of an antibody to an antigen. The “secondary” is generally any antibody that is used to bind to the primary for detection. Secondary antibodies would have alkaline phosphatase (AP) or HRP conjugated to them.
Direct ELISA
A direct ELISA is where the antigen is directly bound to the microplate surface and a directly conjugated antibody is then added for binding/detection.
In brief, the first step of the protocol is to immobilize the antigen to the microplate. The second step is to add a blocking agent to prevent nonspecific binding by the detection antibody. Next, the detection antibody, which is directly conjugated to an enzyme (such as HRP), is added. The detection antibody will bind to the antigen, and the excess is washed out of the well. A substrate for the enzyme is then added and, after a short incubation, the signal from each well would typically be measured on a plate reader.
A direct ELISA has few protocol steps, translating to an overall easy assay set-up. One disadvantage is the necessity of conjugating each antibody that will be used for detection in each assay. Also, the coated antigen is nonspecifically bound to the surface, so it may be presented to the antibody in a non-native configuration, leading to a higher background signal. This assay method is not easily amenable to multiplexing. Generally, the format is ideal for measuring the amount of antibody in samples, such as the serum level of antibody in response to an infection.
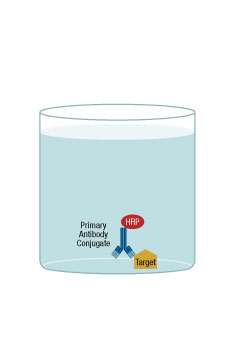
ELISA Direct
Indirect ELISA
The indirect ELISA is similar to the direct ELISA; however, a major difference is that the binding antibody is detected through a second, conjugated antibody. The first step of the protocol is to immobilize the antigen to the microplate. The second step is to add a blocking agent to prevent non-specific binding by the binding antibody. Third, an antigen-specific binding antibody is added and, after brief incubation, the well is washed with buffer to remove the excess reagent. The next step is the “indirect” detection, where an antibody is added to detect the first binding antibody. This antibody is typically conjugated to an enzyme. After incubation and washing the well, substrate for the enzyme is added. Then, after a short incubation, the signal from each well would typically be measured on a plate reader. The second antibody would typically be an antispecies antibody.
One variation of this scheme is to have the detection antibody conjugated to biotin. After antibody addition, a streptavidin-enzyme conjugate can be added and the rest of the protocol continues as before
The major advantage of an indirect ELISA is that the conjugated second antibody can be used for multiple assays, provided the same species of binding antibody is used for all of the different assays. Another advantage is that the second antibody will give an amplification of signal, because about 2 of these antibodies will bind to the first antibody. With an increase in the number of steps, the protocol typically will take longer and could be more prone to error. Additionally there may be an increase in the background signal because of the greater number of reagents required, each one with a potential for nonspecific binding.
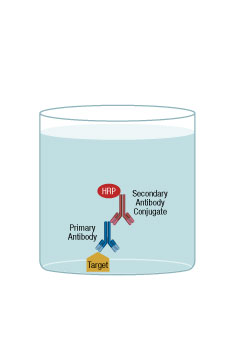
ELISA Indirect
Sandwich ELISA
In a sandwich ELISA, the antigen becomes the center of a “sandwich” between 2 antibodies. One form of sandwich ELISA is where the first antibody is adsorbed onto a microplate well and, after washing and blocking, sample containing antigen is added. A second antibody is added that will bind to another site of the sample molecule. This second antibody is typically conjugated to an enzyme so it can be used for detection. After washing the well, substrate for the enzyme is added. Then, after a short reaction incubation, the signal from each well would typically be measured on a plate reader.
As with the direct and indirect ELISA schemes, there are variations on the general sandwich ELISA. On the detection side, instead of a conjugated antibody, a third antibody that is enzyme-conjugated could be added that would recognize the second antibody. For example, a rabbit antibody is used to coat a microplate, it binds to the sample, and a second, mouse antibody is added that also binds to the sample. Then, an enzyme-conjugated antibody that recognized the mouse antibody is added. A further extension would be to have the second antibody biotin conjugated, which would then require addition of streptavidin-enzyme conjugate.
A further extension of the scheme involves having both antibodies conjugated, one with a tag and one with an enzyme. In this case, there is an anti-tag antibody absorbed to the microplate. The 2 antibodies and the sample are all added to this well together. The sandwich of the tag-conjugated antibody and the sample/enzyme-conjugated antibody will bind to the plate via the anti-tag antibody. This scheme reduces the number of protocol steps, allowing for a less complex, faster assay overall.
In most cases, sandwich assays are more specific because the assay requires 2 antibodies to recognize the target of interest. This specificity requires the development of a “matched set” of antibodies that will work in the assay, a development effort that can be both costly and time consuming.
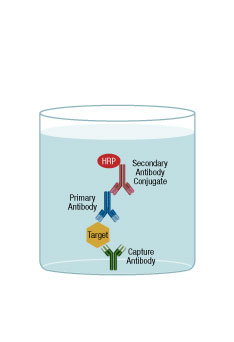
ELISA Capture (Sandwich)
Competition/Inhibition ELISA
Competition/inhibition ELISA, also referred to as blocking ELISA, measures the amount of analyte present by quantifying its interference with an expected signal. One method for doing this is to coat a plate with an antibody specific for a target. The target could be conjugated to an enzyme and used for detection. If the assay contains a sub-maximal amount of detection target, then a high concentration of unlabeled target could compete for binding, lowering the signal. This type of competition assay would be used to determine levels of a specific analyte.
If instead the analyte is coated on the plate, the detection would be performed using antibody conjugated with enzyme. In this case, the amount of antibody in serum could be determined.
This kind of ELISA is useful for measuring immune responses. Also, if a molecule is too small for a sandwich assay, this provides a useful tool for quantitative measurement.
Detection Methods
Detection methodologies for ELISA are many, but the most prevalent in the laboratory are colorimetric, fluorescent, and chemiluminescent. The fundamental aspect of these modalities is that an enzyme, most commonly HRP or AP, is conjugated to the antibody. During the detection step of the protocol, a substrate for these enzymes is added that will form either a chromogenic, fluorescent, or chemiluminescent signal. Because this is an enzymatic process, the reaction can be allowed to progress for a period of time, amplifying the signal, and then stopped. The resulting signal is measured on a plate reader.
The type of substrate used depends on several factors, most notably the desired assay sensitivity and signal to noise ratio.
Colorimetric
The most commonly used type of detection for ELISA assays is colorimetric. Substrates for AP or HRP that create a visible endpoint are robust and stable for long periods of time, making them a good choice for detection chemistries. There are several, and the most common one is 3,3',5,5'-tetramethylbenzidine (TMB).
The amount of color in each well is read by a spectrophotometer, and samples are compared relative to one another or with the use of a standard curve derived from known analyte concentrations.
Fluorescent
Fluorescent substrates for AP and HRP can potentially yield a higher signal and broader dynamic range. Many of these substrates have a shorter half-life than colorimetric substrates, so the signal will drop over time. One more commonly used substrate is 10-Acetyl-3,7-dihydroxyphenoxazine. If instead the analyte is coated on the plate, the detection would be performed using antibody conjugated with enzyme. In this case, the amount of antibody in serum could be determined.
This kind of ELISA is useful for measuring immune responses. Also, if a molecule is too small for a sandwich assay, this provides a useful tool for quantitative measurement.
Chemiluminescent
Detection antibodies conjugated with AP or HRP, for example, can also be used for chemiluminescent assays. In this type of experiment, AP or HRP, in the presence of peroxide, will oxidize luminol, creating light emission at 425 nm. The advantages of this detection type are typically a higher dynamic range and lower background signal leading to increased sensitivity. The signal is not as stable as the other 2 detection types and must be read within a short time of generating the signal.
Components of ELISA
Many kits have been developed to streamline ELISA experiments and to expedite, simplify, and standardize the procedure for each type of ELISA. Reagents have been developed to increase specificity, reproducibility, and sensitivity and to minimize cost. However, when premade kits are not available for the assessment of a particular target of interest, experimenters can prepare the plates and necessary reagents. Below is an overview of the components needed in an ELISA experiment.
Sample Preparation
Numerous types of samples, such as blood, urine, and lysed cells, are routinely analyzed via ELISA. Sample preparation will depend on the particular question being asked. Measurement of secreted proteins in serum, such as cytokines, may be used directly or, in some cases, require minimal clean-up steps. At the other end of the spectrum are measurement of post-translational modifications, which generally require cell lysis in buffers that include protease/phosphatase inhibitors. Since sample preparation can vastly alter the experiment and experimental results, this needs to be carefully considered.
Buffers
Several buffers are needed for an ELISA experiment. These include coating, blocking, washing, dilution, capture, and detection buffers. Basic recipes for these buffers are widely available; however, most are also commercially available, delivering savings in time and providing enhanced quality control to the researcher:
- Coating buffer – This buffer is needed to promote the passive adsorption of an antigen or antibody to the surface of a well by stabilizing the antigen or antibody. The adsorption and immobilization occur as a result of interactions between the antigen or coating antibody and the plastic well. Several factors influence this passive binding, including surface chemistry of the plastic, temperature, pH of the coating buffer, antigen/antibody concentration, and time. Typical coating buffers include (phosphate-buffered saline (PBS), sodium bicarbonate, or similar buffers, but these conditions should be tested and optimized. Importantly, coating buffers should not contain proteins that can compete with the binding of the antigen or antibody.
- Blocking buffer – This buffer prevents the nonspecific binding of detection antibodies to the plate surface. The buffer will either contain proteins to block binding sites or detergents that will prevent binding. A protein-based blocking buffer can only be added after the coating step, because proteins in the solution become adsorbed onto all unbound sites on the plastic. Like the coated analyte or antibody, these nonspecific proteins are not removed through any washing steps. Blocking buffers need to be optimized to allow for maximal sensitivity but minimal background and should not contain components that will interfere with the detection antibody.
- Washing buffer – These buffers are used between steps to remove any unbound material and need to be fully removed after use. The number of washes, the length of each wash, as well as the amount of detergent in the buffer need to be optimized.
- Detection buffers – These buffers contain the needed substrates for catalysis by the antibody-enzyme complex. Many of these substrates are sensitive to buffer conditions, so when considering a new detection chemistry, the lysis buffer and other additives must be considered to reduce nonspecific cleavage of the detection molecules.
Microplates
ELISAs can be performed in a variety of microplate formats, including 96-well microwell strips and 96-, 384-, and 1536-well plates.
The most common plates have a flat well bottom and are made of polystyrene. Plates are treated in numerous ways to generate hydrophobic/hydrophilic or specific reactive moieties.
For example, protein A may be used in order to achieve a more specific attachment to the surface. Choice of plate and surface coating will translate to decreased background and increased binding.
Detection chemistry often influences plate color choice: clear for chromogenic, black for fluorescent, and white for colorimetric.
Antibodies for ELISA
Antibodies selected for an ELISA should be carefully chosen and validated for use. The success of the experiment and reliability of the results are predicated upon highly specific binding of the chosen antibodies to the antigen of interest. Along with specificity, the chosen antibodies should have high affinity and avidity for the antigen. That is, the antibody should bind to an epitope that is on 1 antigen of a single species (specificity), this binding should occur rapidly and strongly (affinity) and, once bound, the antibody-antigen complex should be stable (avidity).
Antibodies selected for sandwich assays have the added complexity that each antibody needs to be directed at a different epitope. Depending on the experimental needs, the 2 antibodies for sandwich assays can be from different species or the same species.
Both monoclonal and polyclonal antibodies can be used in ELISAs, but each provides certain advantages that can be utilized to the experimenter’s advantage.
Advantages of Monoclonal or Polyclonal Antibodies for ELISA
Monoclonal antibodies:
- specific to a single epitope, so they do not interfere with the binding of other antibodies to other regions of the antigen
- less likely to bind nonspecifically to other antigens
- can be used in all steps of an ELISA
- particularly useful in sandwich ELISAs as part of a matched pair of noncompeting antibodies
Polyclonal antibodies:
- pooled collection of antibodies that recognize different epitopes on the same antigen
- provides more robust signals due to the potential for multiple binding sites on a single target protein
For the antibody that requires labeling with an enzyme, such as HRP or AP, labeling efficiency, lot-to-lot variation, and consistent binding after conjugation are all features that need to be addressed to create a high quality assay.
Controls for ELISA
Controls are necessary to ensure that the signal from each sample is physiologically accurate. A group of controls are used in combination to interrogate the validity of an ELISA experiment. These are as follows:
- Blank controls are needed to provide a reference for background signal and to ensure that the readout is indicative of the concentration of analyte in the sample and not an artifact of the experimental setup.
- Positive and negative controls are needed for comparison with the signal produced by the analyte.
- Spiked controls may be needed to indicate assay performance by giving information about the extent of analyte recovery in a particular sample matrix.
Microplate Reader
The chosen microplate reader should match the type of detection used in an ELISA. Colorimetric reactions are the least sensitive, followed by chemiluminescent and fluorescent reactions. The type of detection used should be determined based on the amount of analyte in the sample and whether amplification and/or multiplexing are needed. Importantly, the dynamic range and sensitivity of detection by a specific microplate reader should also influence the choice of detection method.
How to Optimize ELISA Experiment
Optimization of an ELISA experiment is essential to its success. Since ELISA is a multistep procedure, each component can be individually tested prior to the start of an experiment.
Sample Preparation
Sample preparation needs to be optimized based on starting material. Samples with high concentrations of antibodies need additional processing steps before analysis via ELISA. These samples are said to have “matrix effects” that can interfere with proper binding of a capture or detection antibody due to interactions with the high quantity of proteins or their breakdown products. These effects can be overcome using proper coating, washing, and blocking buffers as well as a high dilution of the initial sample.
Buffers (Coating, Blocking, Lysis/Binding, Wash)
Commercially available, prevalidated buffers should be used when possible. If these are not available, then buffers should be optimized as follows:
- The coating buffer should preserve the biological activity and stability of any proteins, especially bound analytes, without interfering with their passive binding to the plastic or changing their epitopes.
- Blocking buffers should contain the correct concentration of nonionic detergent and/or nonspecific proteins to bind to open sites on the coated plate.
- The sample will either be in a lysis buffer if cells are used or a formulated binding buffer. Care needs to be taken so the components of the lysis buffer will not interfere with the antigen/antibody binding yet still lyse the cells.
- Washing buffers should contain the correct amount of salts and detergents and should be at the correct pH in order to stabilize the antibody-antigen complexes while simultaneously reducing background noise.
Microplates
Consistency across wells is integral for accuracy and reproducibility of an ELISA. This includes proper choice of plate, accuracy in pipetting, and maintenance of temperature, humidity, and pH. For plate coating, optimal coating conditions and plate-binding capacity can vary with each protein and must be determined experimentally.
Antibodies
ELISA is highly dependent on choice of antibody; therefore, any antibody used in capture and direct/indirect detection should be assessed for its specificity, affinity, and avidity and empirically tested and optimized.
Proper titration of antibody concentration is crucial. Serial dilutions of capture and detection antibodies should be carefully prepared. The resulting signal should be compared to proper controls to identify any artifacts in the experiment, as well as to determine the dynamic range of detection. The ideal concentration would provide the highest signal and lowest noise.
In addition, the type of antibody should be chosen carefully. For example, for sandwich ELISA, if monoclonal antibodies are to be used, they must be raised against different epitopes so as to not compete with one another. Based on the type of sample (ie, homogeneous or heterogeneous) and the expected analyte concentration, the choice of polyclonal vs monoclonal antibody should be clear. This choice should then be confirmed and validated experimentally before use in an ELISA.
ELISA Results
ELISA results fall within 3 broad categories: quantitative, semiquantitative, and qualitative.
A quantitative ELISA requires use of standards in order to interpret the results of the experimental samples. A standard curve, typically a serial dilution, of known concentrations of an analyte or other standard is generated. The readouts from the samples are compared to the standard curve, and an absolute quantification of analyte concentration is determined.
If an ELISA is considered to have relative quantitation, or be semiquantitative, the readouts from experimental samples are either compared to one another or to a reference sample ,and the concentration of the analyte is presented relative to those other samples.
A qualitative ELISA is one when the presence of signal, as compared to a blank well or unrelated control, is used to determine the mere presence or absence of an analyte in a sample.
How to Interpret ELISA Results
In general, the readout from each ELISA well is given as a numerical value. The results of an ELISA are displayed as measurements of relative light units (RLU) or relative fluorescent units (RFU) vs the log of analyte concentration.
In order to measure the absolute concentration of analyte in each well, a standard curve constructed from a serial dilution of a known quantity of antigen is needed. Then, the measurement of signal in each experimental sample is compared to the standard curve, and the concentration of analyte is derived. An experimental design that produces a highly sensitive assay is needed for this type of analysis.
If absolute quantification is not needed, then relative quantification can be carried out by comparing samples to each other or to a reference sample. For example, a sample of serum antibodies produced in response to an HIV infection can be compared to a sample of basal serum antibodies, and the ratio of both measurements is indicative of the fold-change in the amount of HIV-triggered antibodies.
Lastly, if quantitative analyses are not needed, then the presence of any signal compared to a blank sample or a negative control would indicate the presence of the analyte. This is the least rigorous type of analysis.
Most Frequent Problem Encountered When Running ELISA Test
High well-to-well or run-to-run variability, no signal, weak signal, and high background are all frequently encountered issues in an ELISA. The previously described optimization steps can help with troubleshooting these problems.
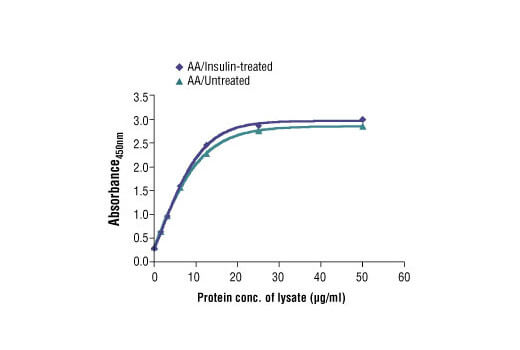
PathScan® Total 4E-BP1 Sandwich ELISA Kit #7179: The relationship between the protein concentration of the lysate from amino acid (AA)/untreated and AA/insulin-treated HEK-293T cells and the absorbance at 450 nm is shown.