Cellular Senescence
What Is Senescence?
Cellular senescence refers to a state of stable cell cycle arrest in which proliferating cells become resistant to growth-promoting stimuli, typically in response to DNA damage. Senescence was first described by Leonard Hayflick upon the observation that human fetal fibroblasts eventually stopped dividing, but remained viable and metabolically active after prolonged time in culture. It is now generally accepted that only transformed malignant cells replicate indefinitely, while non-transformed cells do not, with the exception of cell types with stem-like properties. These include endogenous germline and somatic stem cells, in addition to embryonic or induced pluripotent stem cells developed under controlled in vitro conditions. Senescent cells are distinct from both quiescent cells which can reenter the cell cycle and from terminally differentiated cells.
Senescent cells are characterized by morphological and metabolic changes, chromatin reorganization, altered gene expression, and adoption of a pro-inflammatory phenotype known as the senescence-associated secretory phenotype (SASP). The biological role of senescence is complex as both protective and deleterious effects of senescent cells have been described, largely dependent upon physiological context. For example, while senescence has likely evolved as a mechanism to avoid malignant transformation of damaged cells, the onset of senescence may contribute to many age-associated pathologies, including cancer, tissue degeneration and inflammatory diseases.
The terms aging and cellular senescence cannot be used interchangeably. Aging is a progressive decline with time whereas senescence occurs throughout the lifespan, including during embryogenesis. The number of senescent cells increases with age, but senescence also plays an important role during development as well as during wound healing.
β-Galactosidase staining detects expression of pH-dependent β-galactosidase activity (cells stained in blue), a known characteristic of senescent cells. Normal cells (left panel); Senescent cells (right panel).
Why Does Senescence Occur?
Senescence prevents the replication of cells harboring damaged DNA, which serves an important anti-tumorigenic function. Senescence typically occurs in response to damaging stimuli, including telomere shortening (replicative senescence), DNA damage (DNA damage-induced senescence), and oncogenic signaling (oncogene-induced senescence).
Understanding Replicative Senescence and Hayflick Limit
Replicative senescence refers to the phenomenon whereby normal nonmalignant cells stop dividing in vitro, after approximately fifty divisions, which has been termed the Hayflick limit. Replicative senescence is induced by telomere shortening. With each round of DNA replication, telomeres are progressively shortened, eventually reaching a critical length which prevents further replication, thereby halting cell division. Critically short, uncapped telomeres initiate a DNA damage response which triggers senescence.
DNA Damage-Induced Senescence
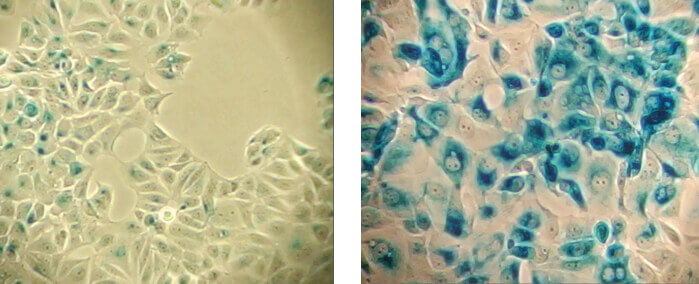
DNA damage triggers the DNA repair machinery, apoptosis, or senescence depending on the extent of damage and physiological context. Senescent cells are characterized by a persistent DNA damage response (DDR), including chronic ATM (Ataxia Telangiectasia mutated) and ATR (Ataxia Telangiectasia and Rad3 related) kinase signaling which ultimately invokes cell cycle arrest and senescence through activation of the p53/p21 and p16/pRb pathways. Persistent DNA damage and subsequent senescence can also be induced by ionizing radiation, chemotherapeutics, genotoxic stress, and oxidative stress.
Cell Cycle G2/M DNA Damage Signaling
Oncogene-Induced Senescence
Cellular senescence is induced in response to oncogenic signaling as a potent cell autonomous anti-cancer mechanism. Senescence occurring in cells with oncogenic signaling is a response intended to prevent their transformation to malignant cells. Oncogene-induced senescence (OIS) results from the hyperactivation of oncogenes like H-Ras or the inactivation of tumor suppressors such as PTEN. For example, expression of H-RASV12, an oncogenic form of the GTPase H-RAS, triggers OIS by inducing chronic p38 mitogen-activated protein kinase (p38 MAPK) signaling. Strong mitogenic signaling can also induce DNA damage via replication stress which triggers the collapse of stalled replication forks.
Biomarkers of Senescence
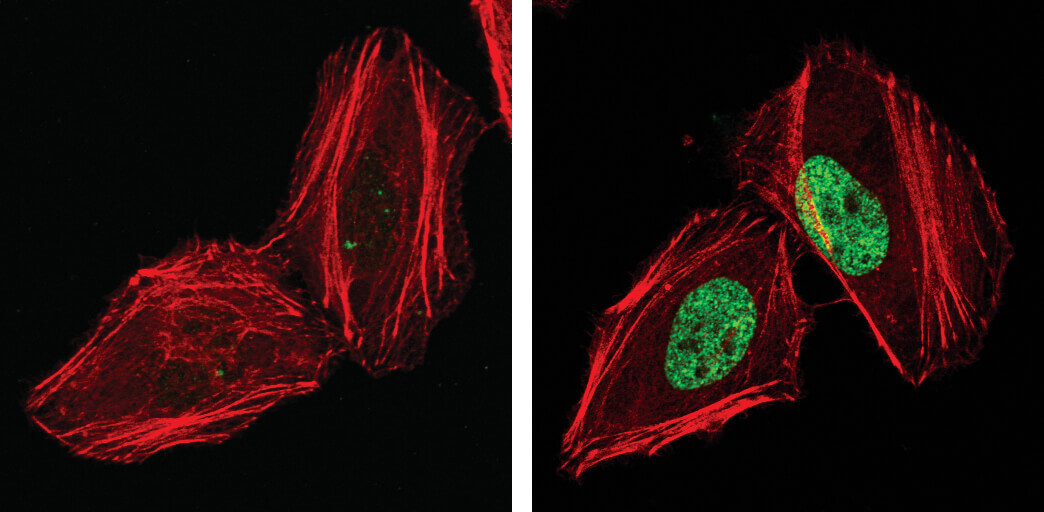
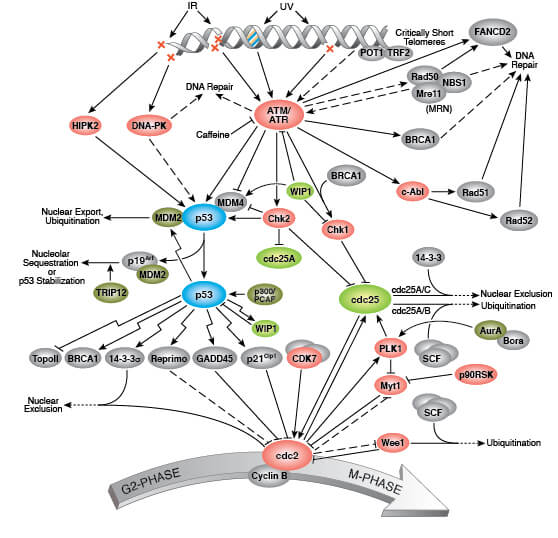
Phospho-Histone H2A.X (Ser139), also known as γ-H2A.X is a commonly used marker of cell senescence (green-stained nucleus in the image on the right).
Senescent cells are characterized by stable cell cycle arrest as well as morphological and metabolic changes, chromatin reorganization, altered gene expression, and acquisition of the senescence-associated secretory phenotype (SASP). It is important to note that not all senescent cells display all biomarkers of senescence. In addition, senescence biomarkers are not necessarily specific to senescent cells, as some markers are observed in apoptotic cells or quiescent cells, for example. Therefore, the identification of senescent cells depends on the observation of several biomarkers, such as those described below:
Stable cell cycle arrest
Only cells with stable cell cycle arrest are considered senescent. Unlike a quiescent cell, a senescent cell will not reenter the cell cycle in response to any known physiological stimuli. Cell cycle arrest is mediated by the p53/p21CIP1 and p16INK4A/pRb tumor suppressor pathways, described in more detail below. Expression of p16INK4A is frequently observed in senescent cells, serving as a useful biomarker. However, p16INK4A is also highly expressed in pRb-negative tumors and cell lines.
Morphological and metabolic changes
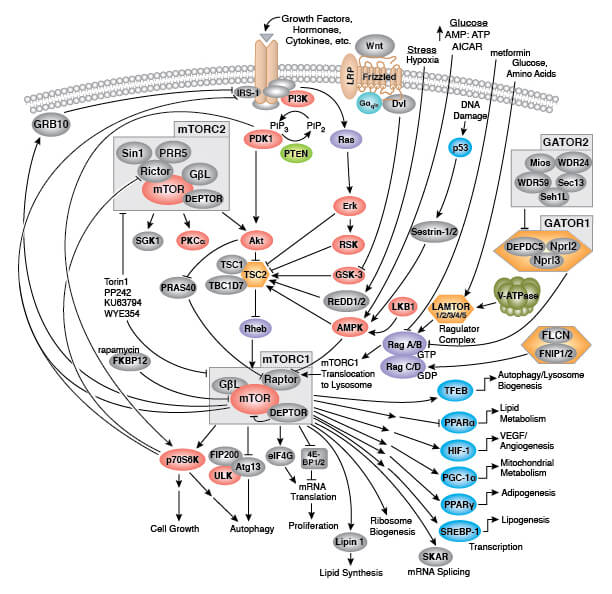
Senescent cells typically have an enlarged size and flattened shape compared to their dividing cell counterparts. Senescent cells display extensive vacuolization and are sometimes multi-nucleated. In addition, disrupted nuclear envelope integrity is observed due to a loss of lamin B1 expression. Senescent cells accumulate dysfunctional mitochondria and display increased levels of reactive oxygen species (ROS). Increased lysosomal content and altered lysosomal activity is also observed, which is reflected by increased levels of β-galactosidase activity at pH 6.0, leading this to be widely adopted as a biomarker of cellular senescence.
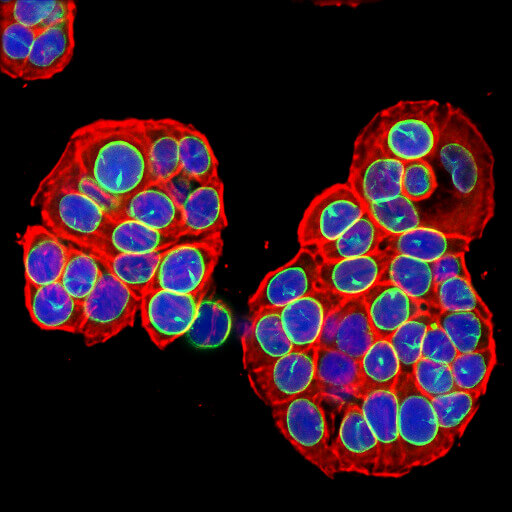
Loss of Lamin B1 (shown here in green) is a marker of cellular senescence.
Chromatin reorganization and altered gene expression
A hallmark feature of senescent cells is extensive chromatin reorganization, most notably the formation of senescence-associated heterochromatin foci (SAHF). These sites of facultative heterochromatin play a role in silencing genes that promote proliferation including E2F target genes like cyclin A. Senescent cells typically contain 30-50 SAHF which are characterized by bright DAPI staining and macroH2A, heterochromatin protein 1 (HP1), and lysine 9 di-or-tri-methylated histone H3 (H3K9Me2/3) immunoreactivity. Although SAHF is frequently observed during senescence, some cells undergo senescence without forming SAHF.
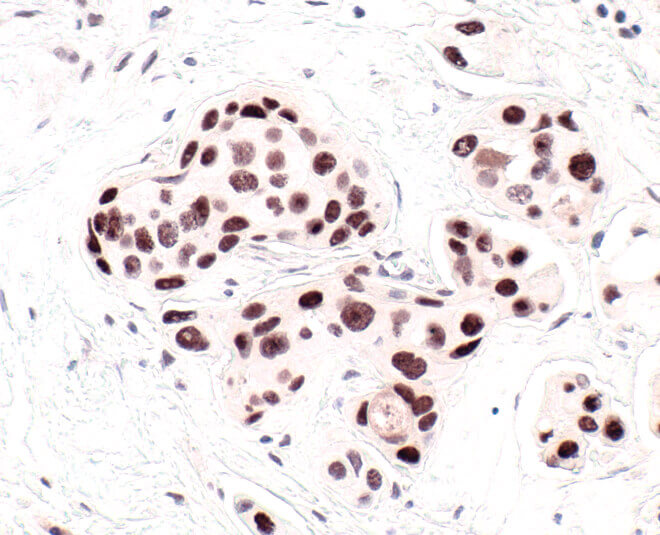
HP1 is a commonly used marker of cellular senescence. Immunohistochemical analysis of HP1 expression in paraffin-embedded human breast carcinoma using HP1 alpha antibody.
DNA damage and persistent DNA damage response (DDR)
DNA damage, such as DNA double strand breaks, is a prominent feature of senescence. Senescent cells display a persistent DNA damage response (DDR) which ultimately triggers cell cycle arrest. Senescent cells contain nuclear foci called DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS), which associate with PML nuclear bodies and accumulate DDR proteins such as activated p53, ATR, and ATM. DNA-SCARS that occur at uncapped telomeres are called telomere dysfunction-induced foci (TIF). Another indicator of DNA damage is γ-H2A.X, which is the phosphorylated form of H2A.X, a variant histone required for checkpoint-mediated cell cycle arrest and DNA repair following double-stranded DNA breaks. DNA damage, caused by ionizing radiation, UV-light, or radiomimetic agents, results in rapid phosphorylation of H2A.X at Ser139.
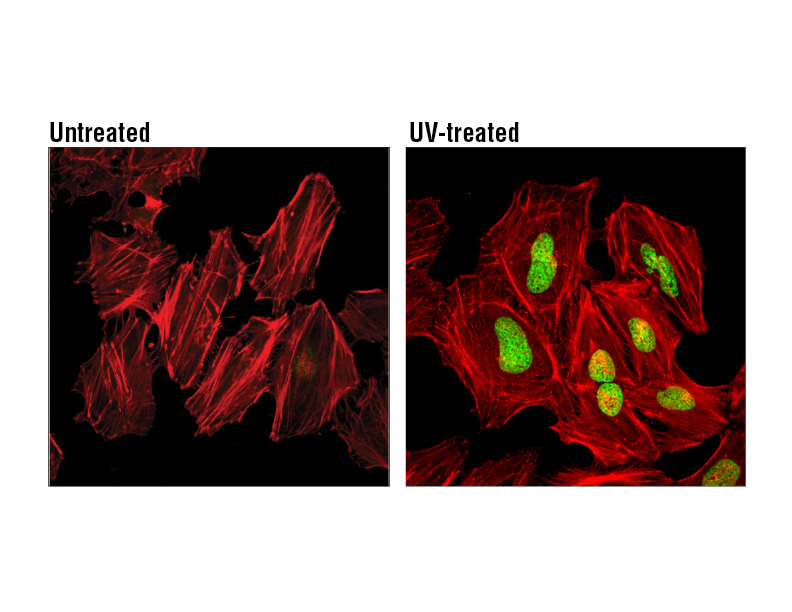
H2A.X is a commonly used marker of cell senescence. Confocal immunofluorescent analysis of HeLa cells, untreated (left), treated with UV (100 mJ/cm2) using Phospho-Histone H2A.X (Ser139) Mouse mAb (green). Actin filaments were labeled with DyLight 554 Phalloidin (red).
Senescence-associated secretory phenotype (SASP)
Many senescent cells acquire a pro-inflammatory senescence-associated secretory phenotype (SASP) that mediates non-cell autonomous effects of senescence, both beneficial and deleterious. The SASP is comprised of a highly complex mixture of secreted cytokines, chemokines, growth factors, and proteases, with the precise composition varying markedly by cell and tissue context and the senescence-inducing stimulus. These secreted factors facilitate communication with neighboring cells and the immune system, which ultimately influences the fate of the senescent cell. For example, the SASP recruits immune cells to senescent cells, thereby facilitating their elimination, which serves a tumor suppressor function. Paradoxically, however, the SASP has been shown to promote tumor cell progression through secretion of factors that promote angiogenesis, extracellular matrix remodeling, or epithelial-mesenchymal transition (EMT). Additionally, chronic senescence-induced inflammation can induce systemic immunosuppression, potentially leading to the onset of diseases including cancer. This chronic inflammation may also drive tissue damage and degeneration associated with aging.
Signaling Pathways for Senescence
Triggers of senescence ultimately converge on the p53/p21CIP1 and p16INK4A/pRb tumor suppressor pathways which induce cell cycle arrest.
Cell Cycle Arrest
Stable cell cycle arrest is a defining feature of senescence, and occurs in response to telomere shortening, oncogenic signaling, and DNA damage. Cell cycle arrest is mediated by the activation of either the p53/p21CIP1 or p16INK4A/phospho-Rb tumor suppressor pathways. Interestingly, mutations in pathway components are commonly observed in different types of cancer.
Cell Cycle G2/M DNA Damage Signaling
Persistent DNA damage response (DDR) due to chronic genomic stress or telomere attrition leads to activation of the tumor suppressor p53, which in turn activates p21CIP1 to initiate cell cycle arrest. Upstream of p53 are the kinases ATR and ATM, which act through Chk1 and Chk2, respectively to activate p53 as part of the DDR. Once activated by p53, p21CIP1 inhibits cyclin dependent kinases (CDKs) which prevents cell cycle progression through the G1 to S phase checkpoint. In addition, CDK inhibition blocks phosphorylation of Retinoblastoma tumor suppressor protein (Rb) which prevents E2F-mediated transcription of proliferation-promoting genes. Inhibition of phosphorylated Rb is also directly mediated by the activation of p16INK4A which prevents CDK4 and CDK6 mediated phosphorylation of Rb and downstream E2F-mediated transcription.
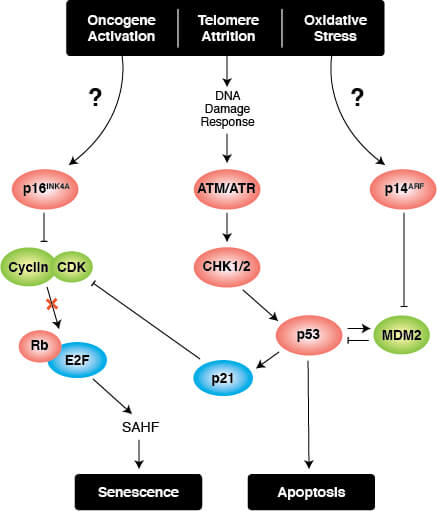
Transcriptional Regulation of Cellular Senescence
Most Common Proteins in Senescence
Antibodies and detection reagents for identifying senescent cells are available from Cell Signaling Technologies:
| Protein or Marker | Role in Senescence |
|---|---|
| Senescence-associated β-galactosidase | Increased activity at pH 6.0 in senescent cells |
| p53 | Activation can trigger cell cycle arrest |
| Rb (Retinoblastoma tumor suppressor protein) | Inhibition triggers cell cycle arrest |
| p21CIP1 | Inhibits cyclin dependent kinases; downstream of p53 |
| p16INK4A | Inhibits phosphorylation and inactivation of pRb |
| Bcl-2 | Increased expression in senescent cells, inhibits apoptosis |
| macroH2A1.1 | macroH2A1 isoform; marker of SAHF |
| macroH2A1.2 | macroH2A1 isoform; marker of SAHF |
| H3K9Me2/3 (lysine 9 di-or-tri-methylated histone H3) | Marker of SAHF |
| HP1 (heterochromatin protein 1) | Marker of SAHF |
| Phospho-Histone H2A.X (Ser139) | Marker of DNA damage |
| Lamin B1 | Expression reduced in senescent cells leading to disruption of nuclear envelope |
| HMGB1 (High mobility group protein B1) | SASP component |
| IL-6 (Interleukin 6) | SASP component |
| TNF-α (Tumor necrosis factor α) | SASP component |
| MMP3 (Matrix metalloproteinase-3) | SASP component |
Telomeres and Their Role in Senescence
Telomere shortening triggers replicative senescence of most somatic cells, which unlike transformed cells, do not express telomerase.
A Brief Explanation of Telomeres and Telomerase
Telomeres are the repetitive nucleotide sequences and associated proteins at the ends of chromosomes that serve as protective caps against chromosomal degradation and end-to-end fusion. Telomeres are comprised of hundreds to thousands of short GT-rich tandem repeat sequences, followed by a single-stranded GT-rich overhang. Telomere-binding proteins constituting the shelterin complex bind the repeat sequence and promote a T-loop structure which caps the chromosome end. The shelterin complex is comprised of telomere repeat factor 1 (TRF1), telomere repeat factor 2 (TRF2), telomeric repeat-binding factor 2-interacting protein (TERF2IP, also known as RAP1), protection of telomere 1 (POT1), TRF1- and TRF2-interacting nuclear protein 2 (TIN2), and the TIN2 binding protein TPP1.
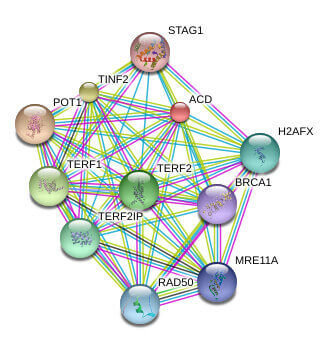
Known and Predicted Protein-Protein Interactions of TPP1
Telomeric sequences cannot be completely replicated by DNA polymerase, which leads to their progressive truncation in most somatic cells. Transformed cells, stem cells, and germ cells express telomerase, a ribonucleoprotein complex that synthesizes telomeric end sequences de novo. The telomerase complex is comprised of telomerase reverse transcriptase (TERT), telomerase RNA (TERC), and dyskerin (DKC1) Because telomere length is maintained in telomerase expressing cells, they are able to continuously proliferate without achieving replicative senescence.
Abnormalities In Telomere Control In Senescence
Disruption of mechanisms controlling telomere stability or length, such as mutations in proteins that form the shelterin or telomerase complex, are associated with genomic instability, tissue damage, and cancer. Genetic disorders associated with defects in the telomere maintenance machinery are collectively referred to as telomeropathies. For example, mutations in the genes encoding TERT, TERC, DKC1, or TINF2 cause dyskeratosis congenita (DKC), a disorder characterized by nail dystrophy, skin hyperpigmentation, and white patches in the mouth (oral leukoplakia). Individuals with DKC frequently develop bone marrow dysfunction and are at a higher risk of leukemia and other cancers. A more severe form of DKC, Hoyeraal-Hreidersson syndrome, causes cerebellar atrophy and microcephaly. Telomere dysfunction is also associated with idiopathic pulmonary fibrosis, familial liver cirrhosis, and spontaneous acute myelogenous leukemia (AML).
Telomere Length and Impact On Senescence
With each round of DNA replication, telomeres are progressively shortened by 50-100 base pairs leading to replicative senescence. During DNA synthesis, DNA polymerase is unable to fully replicate the end sequences on the lagging strand which is known as the “end replication problem”. As a result, these unreplicated sequences are truncated resulting in progressive telomere attrition. Once telomeres reach a critical length, the protective telomeric cap structure is disrupted and recognized by the cell as a DNA double strand break, which triggers a persistent DNA damage response and halts further cell division. Cells that express telomerase, such as cancerous cells, are able to maintain telomere length and avoid senescence.
Why Is Senescence Research Important?
Senescent cells accumulate with age and contribute to the normal aging process as well as age-related disorders. The link between senescence, aging, and age-related pathologies, including cancer, neurodegeneration, and metabolic and cardiovascular diseases have largely fueled the senescence research field. Research in rodent models has shown that selectively eliminating senescent cells in vivo can reduce inflammation, enhance immune system function, and thereby slow the progression of age-related diseases, leading to increased health and lifespan. For example, drugs that induce senescence, including some chemotherapeutics, are effective against cancer by suppressing their replicative potential. However, senescent cells accumulate in patients undergoing chemotherapy, presumably due to DNA damage, and are thought to contribute to unwanted side effects, particularly fatigue. Moreover, senescent cells can also contribute to cancer relapse and metastasis through the release of SASP components. Thus, the use of senolytics, which are targeted therapeutic agents against senescent cells, in chemotherapy patients may help prevent cancer relapse and alleviate some side effects. Senolytic therapies can also extend lifespan and delay age-associated physical decline in normal mice, suggesting they may be effective in treating age-related disorders. Senolytic drugs are currently being tested in humans in clinical trials for treatment of osteoarthritis and chronic kidney disease.
Senescence Research Models
Disorders that cause a condition resembling premature aging, known as progeroid syndromes, have shed light on the role of genomic integrity and DNA damage in the aging process. Such disorders occurring in humans include Werner syndrome, Hutchinson-Gilford progeria, and Xeroderma pigmentosum. Mouse models of progeroid syndromes are being used to study mechanisms underlying premature aging and senescence. For example, mice expressing low levels of BubR1, a cell cycle checkpoint kinase, display accelerated aging and express p16INK4A at high levels in some tissues with age-related pathologies. Germline deletion of p16INK4A delayed aging of BubR1 progeroid mice, which was associated with reduced levels of senescent cells, demonstrating a functional in vivo link between biological aging and cellular senescence. In addition, a transgenic mouse model, INK-ATTAC, in which p16INK4A expressing senescent cells can be selectively eliminated, has shown that the loss of senescent cells increases lifespan and health span of mice.
Several strains of the senescence-accelerated mouse (SAM) model have been developed. These lines were originally derived from mice with a phenotype consistent with accelerated senescence, including decreased activity, hair loss, and shortened lifespan. SAM mice display pathobiological phenotypes such as learning and memory deficits, osteoporosis, and lymphoma.
On the flip side, animal models have also been used to study delayed aging and senescence. For example, the naked mole rat is a small mouse-sized rodent that can live 30 years or more, well beyond the life span expectancy based on their size. Naked mole rats rarely develop cancer and remain healthy and active even at advanced ages. Interestingly, senescence occurs normally in these animals, indicating that longevity does not depend on the elimination of the cellular senescence response.

Naked Mole Rat
Elucidating mechanisms underlying senescence and aging have important implications for understanding, and potentially treating or preventing age-associated illnesses in humans. Metformin, an approved diabetes drug, reduces insulin levels and decreases IGF-1 signaling, leading to AMPK activation and mechanistic target of rapamycin (mTOR) inhibition. Metformin can inhibit the SASP, and in humans may reduce cancer incidence. The Targeting Aging with Metformin (TAME) clinical study will test whether metformin treatment delays or prevents onset of age-related diseases, such as cancer, heart disease, and dementia.