
Protein Kinases
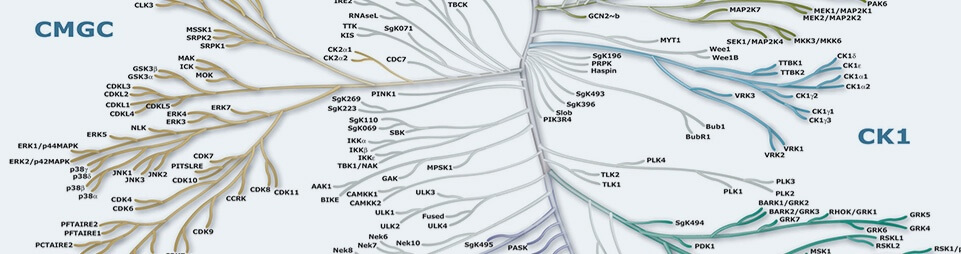
Protein Kinases are key regulators of cell function that constitute one of the largest and most functionally diverse gene families. By adding phosphate groups to substrate proteins, they direct the activity, localization and overall function of many proteins, and serve to orchestrate the activity of almost all cellular processes. Kinases are particularly prominent in signal transduction and co-ordination of complex functions such as the cell cycle. The diversity of essential functions mediated by kinases is shown by the conservation of some 50 distinct kinase families between yeast, invertebrate and mammalian kinomes. Of the 518 human protein kinases, 478 belong to a single superfamily whose catalytic domains are related in sequence. These can be clustered into groups, families and sub-families, of increasing sequence similarity and biochemical function. The kinase dendrograms (above) show the sequence similarity between these catalytic domains: the distance along the branches between two kinases is proportional to the divergence between their sequences. Seven major groups are labeled and colored distinctly. For instance, the tyrosine kinases form a distinct group, whose members phosphorylate proteins on tyrosine residues, whereas enzymes in all other groups phosphorylate primarily serine and threonine residues. The relationships shown on the tree can in some instances be used to predict protein substrates and biological function for many of the over 100 uncharacterized kinases presented here. A further 40 ‘atypical’ kinases have no sequence similarity to typical kinases, but are known or predicted to have enzymatic activity, and some are predicted to have a similar structural fold to typical kinases.
Gerard Manning, Sugen, Inc., South San Francisco, CA, USA