

Synopsis of Cell Proliferation, Metabolic Status, and Cell Death
What Are the Indicators of Cell Viability?
Cell viability is a measure of the proportion of live, healthy cells within a population. Cell viability assays are used to determine the overall health of cells, optimize culture or experimental conditions, and to measure cell survival following treatment with compounds, such as during a drug screen.
Typically, cell viability assays provide a readout of cell health through measurement of metabolic activity, ATP content, or cell proliferation. Cell viability can also be assessed using cell toxicity assays that provide a readout on markers of cell death, such as a loss of membrane integrity.
Together, cell viability and cell toxicity assays are important tools for assessing cellular responses to experimental compounds of interest.
Role of Cell Proliferation as an Indicator of Cell Viability
Cell proliferation assays can measure cell division events directly (counting cells) or use markers of cell division/DNA replication such as DNA content.
What is Cell Proliferation
Cell proliferation refers to an increase in cell number due to cell division (cytokinesis), which occurs as the final step of the cell cycle. Healthy cells actively proliferate whereas growth-arrested, senescent, and dead or dying cells do not. Thus, cell proliferation assays are a useful tool for assessing cell viability or cell survival by providing a readout on the number of actively dividing cells present in a sample.
How to Measure Cell Proliferation
Cell proliferation assays typically measure DNA content or DNA synthesis in replicating cells. Cell proliferation assays are performed using standard methods, including enzyme-linked immunosorbent assay (ELISA), flow cytometry, immunofluorescence and high content imaging.
Cell cycle assay—analysis of DNA content
Cell cycle assays are used to determine the proportion of cells at different stages of the cell cycle via flow cytometry. During cell cycle progression, cells increase in size (G1 phase), which is followed by DNA synthesis and replication (S phase), further growth (G2 phase), and finally by mitosis (M phase) and cell division. Cells that exit the cell cycle and stop dividing remain at resting state (G0) and are referred to as quiescent.
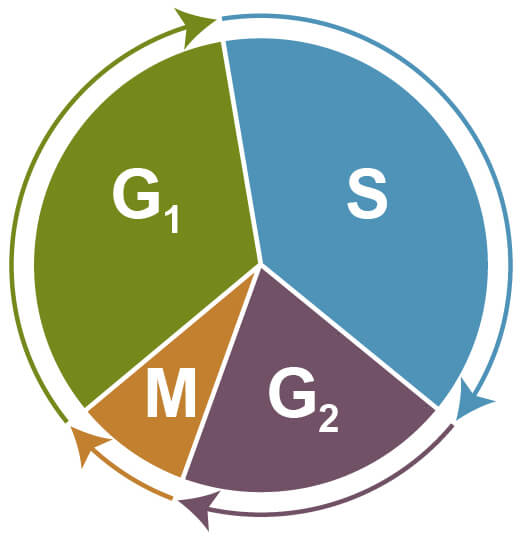
Phases of the Cell Cycle. G1 - increasing size; S - DNA synthesis and replication; G2 - growth; M - mitosis and cell division; G0 - quiescent.
For the cell cycle assay, cells are fixed or permeabilized, incubated with a DNA intercalating agent such as propidium iodide (PI) and subsequently analyzed by flow cytometry. Differences in fluorescence intensity of the PI signal correlate to different phases of the cell cycle. For example, cells in G2 or M phase have twice as much DNA content as cells in G0 or G1 phase, which corresponds to an increased fluorescence signal. Cells in S phase have varying fluorescence intensities which are between that observed for cells in G0/G1 and G2/M phase. Fluorescence signals below the G0/G1 peak typically correspond to apoptotic cells with fragmented DNA.
Proliferation assay—analysis of DNA synthesis
During S phase, nucleoside labeling agents such as 3H-thymidine or 5-bromo-2'-deoxyuridine (BrdU) are incorporated into newly synthesized DNA. The level of reagent incorporation is proportional to the amount of cell division in the target population. While the 3H-thymidine incorporation assay requires the use of radioactivity, BrdU incorporation is detected using an anti-BrdU antibody in ELISA, flow cytometry, or IF-IC applications and does not require the use of radioisotope labeling.
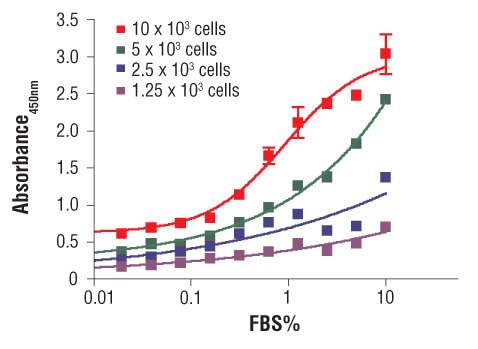
C2C12 cells were seeded at varying density in serum free medium in a 96-well plate and incubated overnight. Serum was added to the plate at various concentrations and cells were incubated for 24 hr. Finally, 10 μM BrdU was added to the plate and cells were incubated for 4 hr
Detection of proliferation proteins
Dividing cells have high expression of cell cycle proteins compared to quiescent or senescent cells, thus the level of cell cycle-specific proteins can be measured as a readout of cell proliferation. Some common proliferation proteins include proliferating cell nuclear antigen (PCNA), Ki67, and Phospho-histone H3, which can be detected using western blot (WB), IF, IHC, flow cytometry, and ELISA.
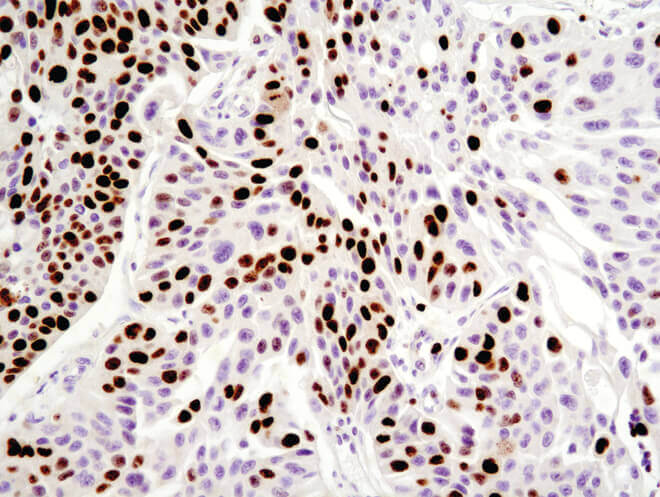
Immunohistochemical analysis of paraffin-embedded human breast carcinoma using Ki-67 (8D5) Mouse mAb.
Senescence to indicate impaired proliferation
Dysfunctional replication can result in senescence, and senescent cells will not divide in response to growth signals. Markers of senescence can therefore be used as a complement to cell proliferation assays. An enlarged cell size, expression of pH-dependent β-galactosidase activity, and an altered pattern of gene expression further characterize senescent cells and assist assessment of cell proliferation.
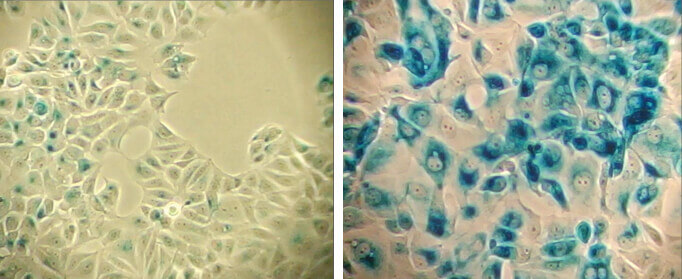
β-Galactosidase staining at pH 6.0 on MCF-7 cells untreated (left) and senescent MCF-7 cells treated with etoposide #2200 (12.5 μM, 24 hr) and allowed to recover for 4 days (right).
Cell Viability
Cell viability assays are used to determine the number of healthy cells in a sample. Aside from proliferation assays described above, other cell viability assays report on the overall health of a population, without distinguishing dividing cells from non-dividing cells.
How to Test Cell Viability
The most common readout of cell viability is with vital dyes such as propidium iodide, however cell viability assays also typically measure the metabolic activity or ATP content of healthy cells.
Metabolic assays such as the MTT and XTT assays quantify cell health by measuring reduction of a colorimetric substrate by mitochondrial enzymes. The MTT assay quantifies the relative quantity of viable cells using this approach. Cultures are incubated with the yellow tetrazolium dye MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) which, in healthy cells, is converted by mitochondrial enzymes into an insoluble purple formazan product. After solubilization by detergents or isopropanol, the number of viable cells can be determined by measuring absorbance at 450 nm in a microplate reader. The XTT cell viability assay is an alternative to the MTT assay which yields a formazan product that is soluble in aqueous solutions, and thus does not require an additional solubilization step.
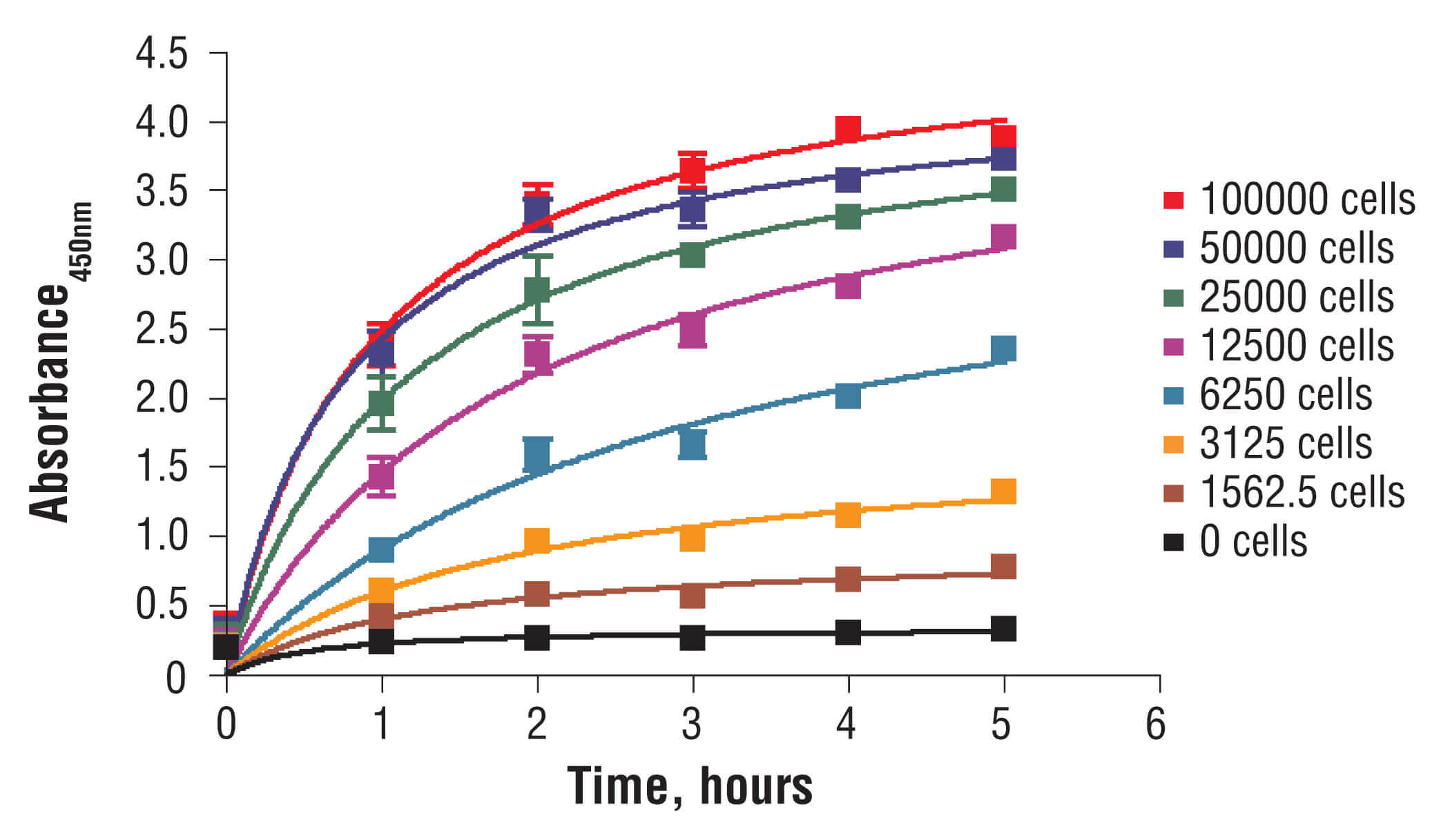
C2C12 cells were seeded at varying density in a 96-well plate and incubated overnight. The XTT assay solution was added to the plate and cells were incubated. The absorbance at 450 nm was measured at 1.0, 2.0, 3.0, 4.0, and 5.0 hours.
ATP measurement assays quantify ATP content to determine the number of viable, metabolically active cells in a sample. These assays are performed in multiwell plates with a colorimetric, fluorometric, or luminescent readout of a metabolic activity requiring ATP, where substrate generation is proportional to the number of healthy cells with active mitochondria.
Cell Toxicity
Cell toxicity refers to the ability of a substance to damage or kill cells. Cell toxicity can be assessed directly by assays that quantify markers of cell death, such as a loss of membrane integrity. Cell toxicity can also be assessed indirectly using the cell viability assays described above.
Cell toxicity assays are useful tools for assessing cell survival following treatment with pharmacological agents or other stressors as well as examining the effect of compounds on immune cell mediated killing of tumor cells using the T cell killing assay.
What is Cell Toxicity?
Cell toxicity refers to the ability of a substance to damage or kill cells.
How to Test Cell Toxicity
Many cell toxicity (cytotoxicity) assays are available that report on different indicators of cell death or compromised cell health.
Live/dead cell counting is used to measure the quantity of both live and dead cells in the sample population. For example, the trypan blue dye exclusion method relies on the principle that only dead or dying cells with damaged membranes take up trypan blue so the number of dead (blue) or viable (colorless) cells can be counted using a hemocytometer.
Cell membrane integrity is assessed to determine if cells have damaged or ruptured membranes due to primary necrosis or secondary necrosis following apoptosis. Membrane damage causes release of cytosolic contents into the extracellular space including the enzyme lactate dehydrogenase (LDH). The amount of extracellular LDH can be analyzed using a colorimetric assay where the amount of product formed correlates to the quantity of dead or damaged cells in the sample. Cell membrane integrity can also be assessed by analyzing the uptake of membrane impermeable dyes such as propidium iodide.
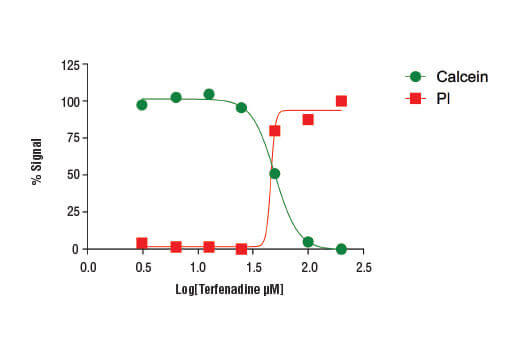
Treatment of HeLa cells (4 x104 cells/well) with increasing concentrations of terfenadine (4 hr) results in reduced cell viability as detected by the Cell Health Assay Kit.
T cell killing assays are used to examine the effect of test compounds on immune cell-mediated killing of cultured tumor cells. Cytotoxic T cells are a class of T cells that recognize and destroy virus-infected cells and tumor cells. In the T cell killing assay, target tumor cells are first labeled with a reporter that permits identification of live versus dead cells. Test compound and cytotoxic T cells are then added to the culture and tumor cell viability monitored using live cell imaging or flow cytometry.
Apoptosis
Apoptosis is a form of programmed cell death necessary for proper growth, development, and homeostasis of multicellular organisms, but can also occur in response to cellular stress. This is different than cell death by necrosis, necroptosis, and pyroptosis. During apoptosis cells undergo characteristic shrinkage, membrane blebbing, nuclear fragmentation, and chromatin condensation. Cell debris is packaged into apoptotic bodies which are then engulfed and digested by phagocytes.
Apoptosis is mediated by a class of proteolytic enzymes called caspases which can be activated via intrinsic (loss of mitochondrial membrane potential and cytochrome c release) or extrinsic (ligand binding to death receptors) apoptotic signaling mechanisms. More detailed information on pathways and regulation of apoptosis is available here.
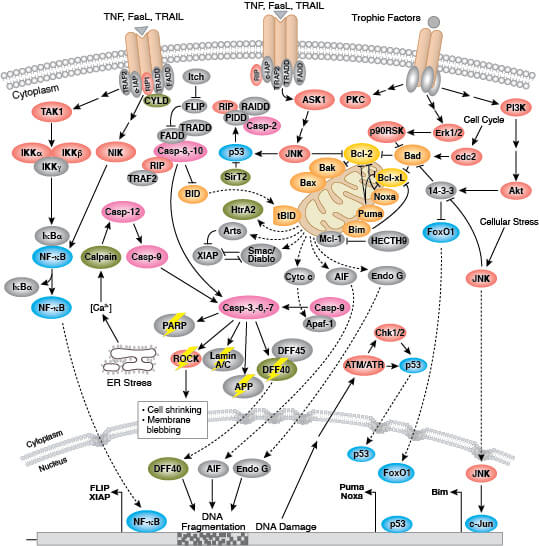
Why Does Cell Apoptosis Occur?
Apoptosis occurs normally during growth and development to eliminate cells that are no longer needed or that are potentially harmful to the organism, such as virus-infected cells or cells harboring damaged DNA. Dysregulation of apoptosis occurs in disease, where elevated levels of apoptosis are associated with autoimmune and neurodegenerative disorders and reduced levels of apoptosis occur in many cancers.
Apoptosis can be induced experimentally by incubating cells with ligands that bind death receptors including TNF-R1 and TNF-R2 or with pro-apoptotic chemical agents like staurosporine or etoposide.
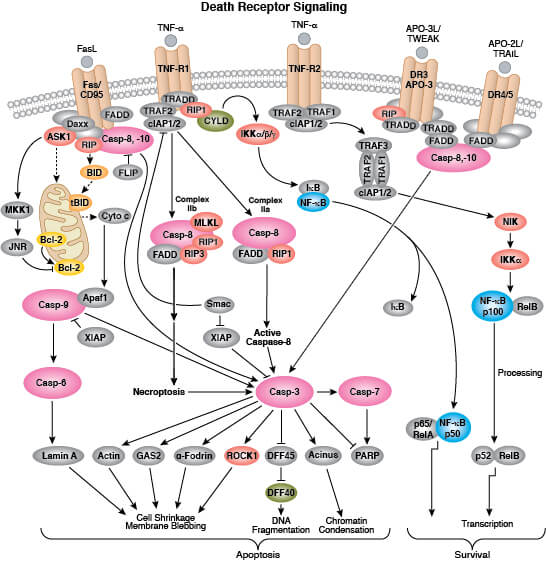
What Is the Difference Between Apoptosis, Necrosis, and Necroptosis/Pyroptosis?
Whereas apoptosis is a form of programmed cell death, necrosis refers to unprogrammed cell death that occurs in response to acute injury or infection. During necrosis, cytoplasm and organelles swell leading to a loss in membrane integrity and ultimately cellular lysis which results in the release of cellular contents into the extracellular space. Necrotic cell death activates an inflammatory response in the surrounding area, whereas apoptotic cell death does not. Secondary necrosis commonly occurs in cell populations undergoing extensive apoptosis.
Although necrosis is typically regarded as an unprogrammed cell death mechanism, a regulated form of necrosis called necroptosis has been described. Necroptosis provides a line of defense against viruses that block the apoptotic machinery during infection by killing infected cells in a caspase-independent manner. This pathway is also activated by pro-inflammatory signaling and ischemic injury.
Pyroptosis is a form of caspase-1 mediated lytic cell death that typically occurs in immune cells as a response to microbial or viral infection.
How to Measure Apoptosis
Apoptosis assays are used to quantify the number of cells undergoing apoptosis. Examples include detection of annexin V binding to exposed membrane lipids, caspase activation, chromatin condensation, DNA fragmentation, or cytochrome c release.
Annexin V assay
Membrane asymmetry, or the irregular distribution of different lipid species on either side of the plasma membrane, is disrupted during apoptosis as phosphatidylserine (PS) translocates from the inner to the outer leaflet of the plasma membrane. The presence of PS on the outer leaflet signals for engulfment and degradation by phagocytes. Exposed PS can be recognized by fluorophore-conjugated annexin V using flow cytometry, IF, or ELISA as an early marker of apoptosis.
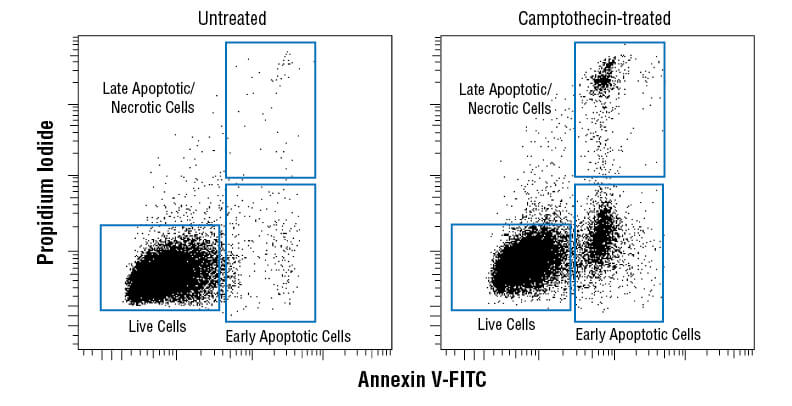
Flow cytometric analysis of Jurkat cells untreated (left) or treated with camptothecin (10μM, 4 hr; right) using Annexin V-FITC Early Apoptosis Detection Kit.
Caspase assay
Caspase enzymes are processed from inactive zymogens to active proteases during the initiation and execution of apoptosis. The initiator caspases-8 and -9 activate executioner caspases like caspase-3. Caspase-3 cleavage can be detected via western blotting, IF, IHC, or flow cytometry as a readout for apoptosis. Caspase-3 activity and caspase substrates like cleaved PARP can also be measured using methods similar to cleaved caspase-3 or by ELISA where the extent of substrate processing is proportional to the quantity of apoptotic cells in the sample. Further examination of apoptotic pathways can be performed using antibodies to other activated caspases.
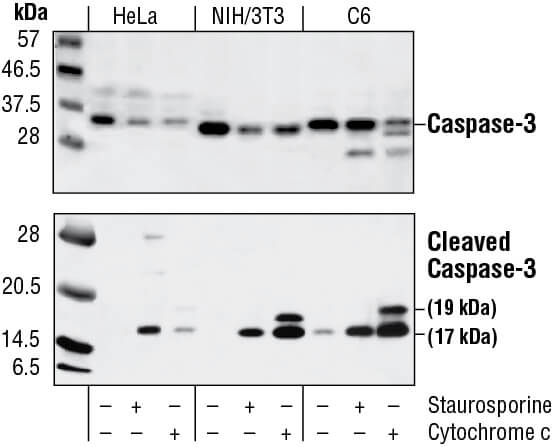
Western blot analysis of extracts from HeLa, NIH/3T3 and C6 cells untreated, staurosporine-treated (3hrs, 1 µM in vivo) or cytochrome c-treated (1hr, 0.25 mg/ml in vitro), using Caspase-3 Antibody (upper) or Cleaved Caspase-3 (Asp175) Antibody (lower).
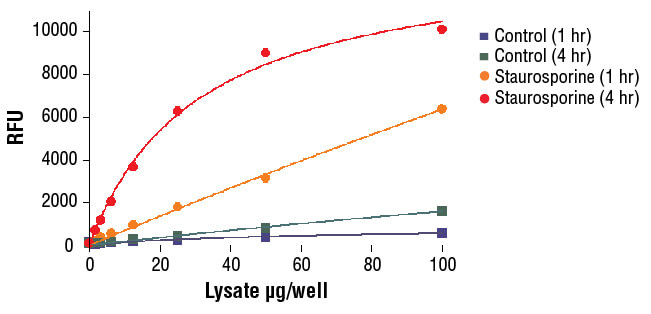
NIH/3T3 cells were treated with Staurosporine #9953 (5 μM, 5 hr) and then lysed in PathScan® Sandwich ELISA Lysis Buffer (1X) #7018 (supplied with kit).
Chromatin condensation
A classic morphological feature of apoptotic cells is the condensation of nuclear chromatin from a heterogeneous to compact state. Chromatin condensation can be observed using nuclear stains and IF or flow cytometry. When stained with nuclear dyes, apoptotic cells have a stronger fluorescent signal due to the presence of condensed chromatin.
TUNEL assay
DNA strand breaks and DNA fragmentation are characteristic of late-stage apoptosis and can be detected using terminal deoxynucleotidyl transferase (TdT) dUTP nick end labeling (TUNEL assay) to quantify the number of apoptotic cells within a population via IF, IHC, plate reader, or flow cytometry. The TdT enzyme catalyzes addition of labeled dUTPs, such as dUTPs conjugated with a fluorophore or biotin, at sites of DNA double strand breaks. Apoptotic cells or other cells with DNA damage are labeled with the modified dUTP while healthy cells are not. Fragmented DNA from apoptotic cells can also be observed using standard gel electrophoresis, as the fragmented DNA will appear laddered compared to the intact DNA from healthy cells.
Cytochrome c release assay
Apoptotic stimuli trigger cytochrome c translocation from the mitochondria to the cytosol. Cellular fractionation can be used to separate the mitochondria from the cytosol, followed by immunoblotting with an antibody against cytochrome c.
How to Measure Necrosis and Necroptosis/Pyroptosis
Necrotic cell death is primarily characterized by a loss of cell membrane integrity, leading to release of cellular contents into the extracellular space, which can be detected using intracellular and secreted markers such as HMGB1, LDH, and IL-1β.
Necroptosis, a genetically programmed form of necrosis, is characterized by the formation of the necrosome, a protein complex containing RIP and RIP3 kinases. Necroptosis can be assayed by measuring the expression or post-translational modification of these and related necroptosis-associated proteins (e.g., MLK1).
In response to microbial infections, the inflammasome forms to initiate caspase 1-dependent pyroptosis. Caspase 1-mediated cleavage results in activation of the proinflammatory cytokines IL-1β and IL-18. Caspase-1 also cleaves gasdermin D, resulting in oligomerization of the N-terminal fragment, forming membrane pores allowing the secretion of IL-1β, IL-18 and other DAMPs.
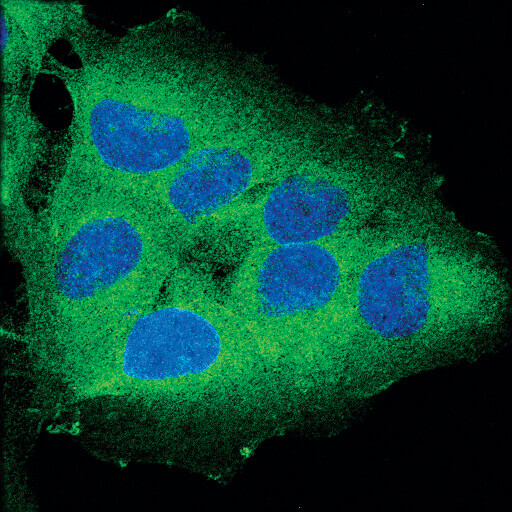
Confocal immunofluorescent analysis of OVCAR8 cells using RIP (D94C12) XP® Rabbit mAb (green). Blue pseudocolor = DRAQ5® #4084 (fluorescent DNA dye).
Choose the Best Method to Assess Cell Survival
The optimal assay for measuring cell survival depends upon many factors, including the experimental context and the cell population under investigation. Multiple, independent assays may be necessary to confirm experimental outcomes.
Considerations for choosing the best method to assess cell survival
Cell type - Experiments conducted in dividing cells are amenable to survival analysis by assays that assess cell proliferation, metabolic activity, or a loss in membrane integrity. For post-mitotic or non-dividing cells like neurons, metabolic or membrane integrity assays are the best choice. Plating density and culture conditions should be standardized for optimal assay performance and to ensure reproducible results.
Timing - The experimental time course should be considered when choosing a cell viability or toxicity assay, as cell survival may be influenced by treatment duration. Additionally, some markers of cell death may be present only at specific stages of the apoptosis process, such as PS translocation, which occurs early in apoptosis, or DNA fragmentation, which occurs late in apoptosis.
Mechanism of cell death - Although standard cell viability and cell toxicity assays will provide quantitative data on live or dead cells in a sample, they do not provide insight into the mechanism of cell death in the experimental context. Targeted experiments using assays that are specific for cell death pathways (e.g. apoptosis) are necessary to elucidate the precise mechanism(s) of cell death.
Assay interpretation - Experimental compounds may not induce cell death, but rather alter cellular metabolism or cellular proliferation, which may incorrectly be interpreted as reduced viability. It is important to use a combination of cell viability and cell toxicity assays to distinguish among these possibilities.
Measuring Cell Viability By Flow Cytometry
While many cell viability assays report on cell health indirectly through measurement of proliferation or metabolic activity, flow cytometry facilitates the direct identification of live and dead cells in a sample. Typically, a membrane-impermeable dye like propidium iodide is used to identify dead or dying cells with damaged membranes and a viability dye like calcein-AM used to label live cells.
Treatment of HeLa cells (4 x104 cells/well) with increasing concentrations of terfenadine (4 hr) results in reduced cell viability as detected by the Cell Health Assay Kit.
Immunohistochemistry (IHC) to Assay Cell Viability
Immunohistochemistry (IHC) uses antibodies to detect cell populations in tissue samples that express specific antigens. This approach is commonly used to identify the temporal distribution of proteins during development, analyze protein expression patterns in disease versus healthy tissue, and to distinguish individual cell types based on biomarker expression. IHC can also be used to measure cell survival in context by analysis of tissue samples. For example, cell viability can be assessed by quantifying the number of cells expressing proliferative markers such as Ki67 or PCNA, which is particularly relevant in determining the effect of treatments on tumor cell proliferation. In addition to the TUNEL assay, apoptotic cells can be identified by staining tissue for antibodies against cleaved caspase-3 and cleaved PARP.
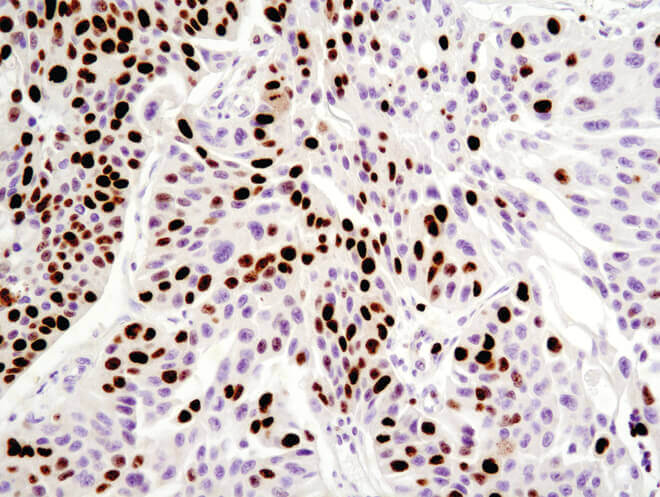
Immunohistochemical analysis of paraffin-embedded human ovarian serous adenocarcinoma using Ki-67 (8D5) Mouse mAb.